¶ Introduction
The Information Browser layout collects general information about the structure, and access structure files and attached files.
It is organized in tabs (SUMMARY, METHODS, LIGANDS, FILES), that sort the metadata intuitively for easier accessibility.
¶ Access
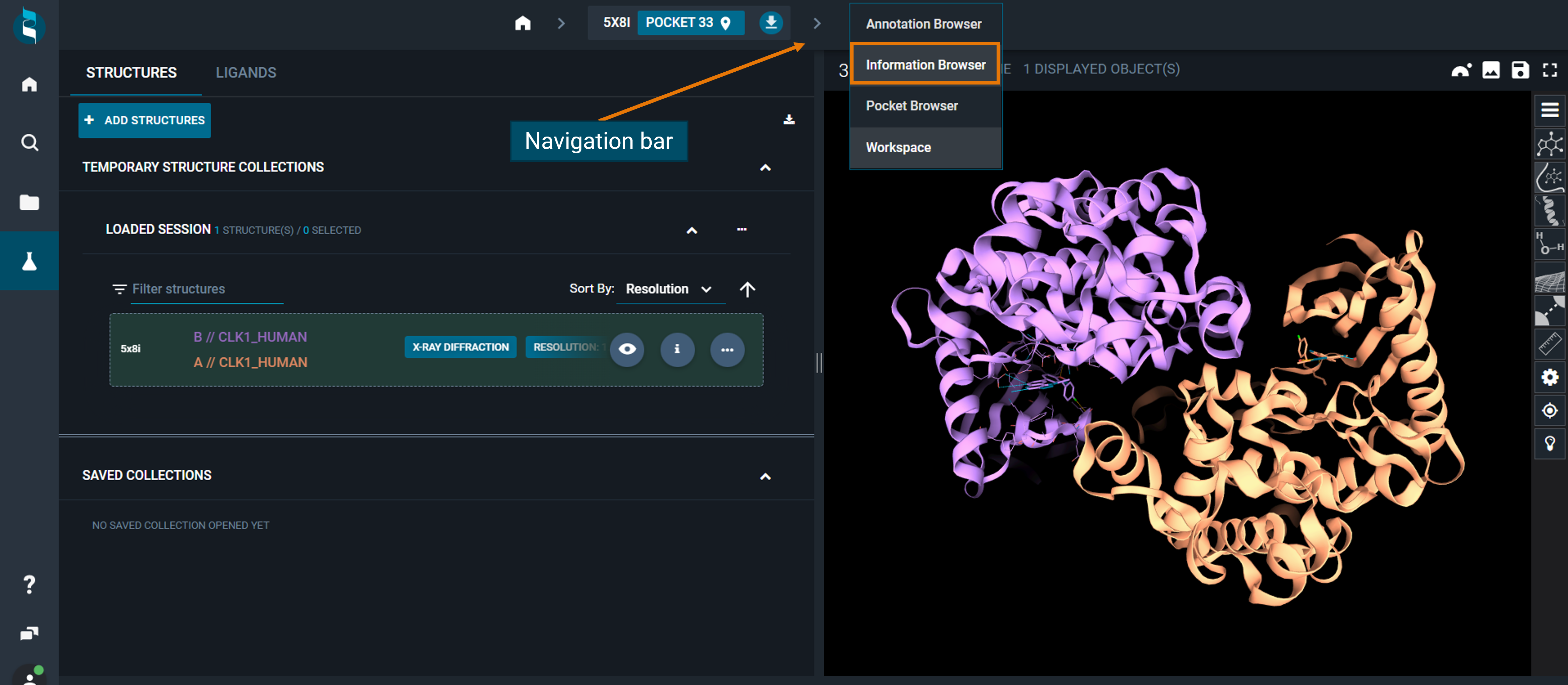
You can access the Information Browser from the Workspace, clicking on the navigation bar on the top, from the drop-down menu. This will give you information on the reference structure in the workspace.
Alternatively, you can have a quick-view of the structure information of any structure loaded in the workspace by clicking on the i button in the structure card. The Information Panel for the specific structure will replace the workspace dashboard.
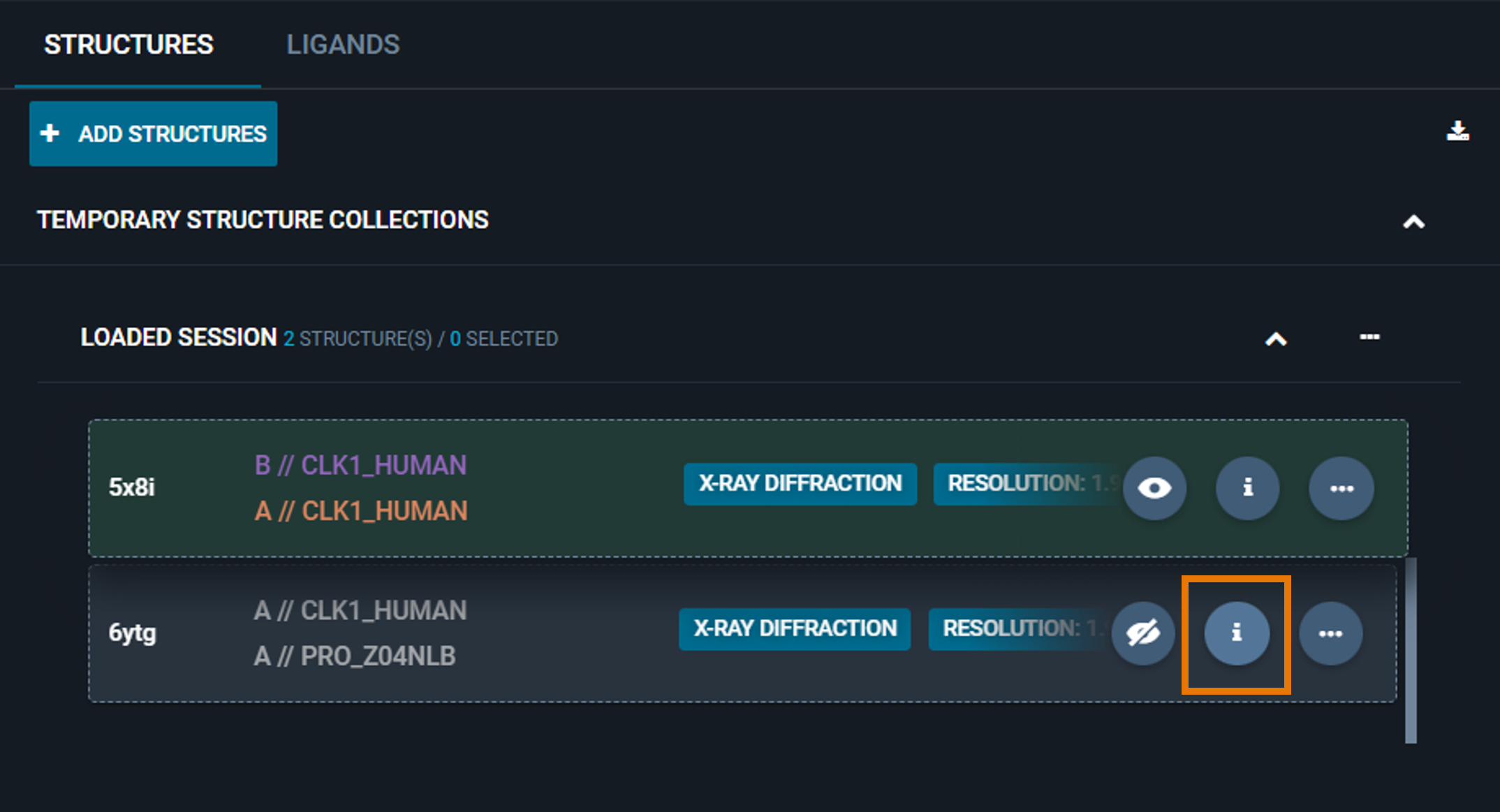
In this case, you can restore the workspace view by clicking on the blue BACK button on the top right.
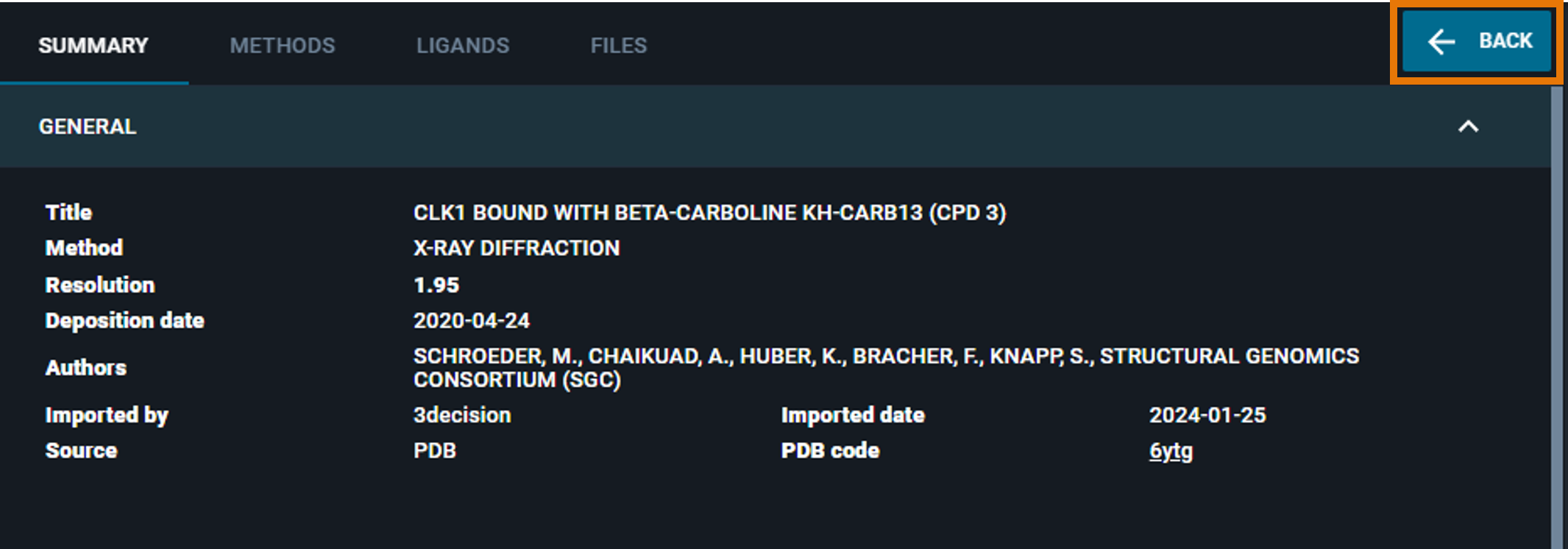
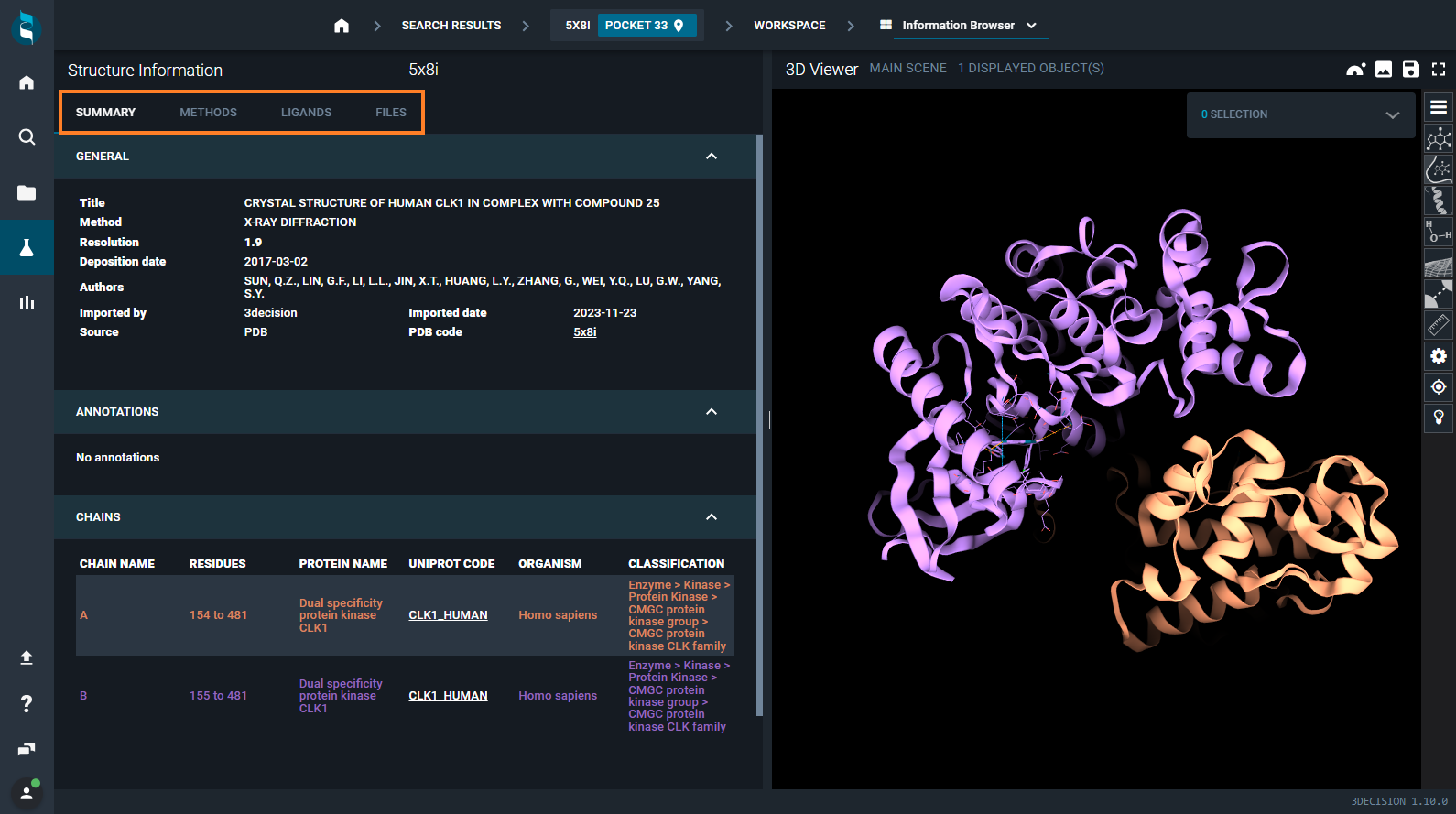
¶ Information Browser
The information are listed on the left part of the view, while on the right the 3D Viewer is kept open. Information are organized in four tabs:
- SUMMARY
- METHODS
- LIGANDS
- FILES
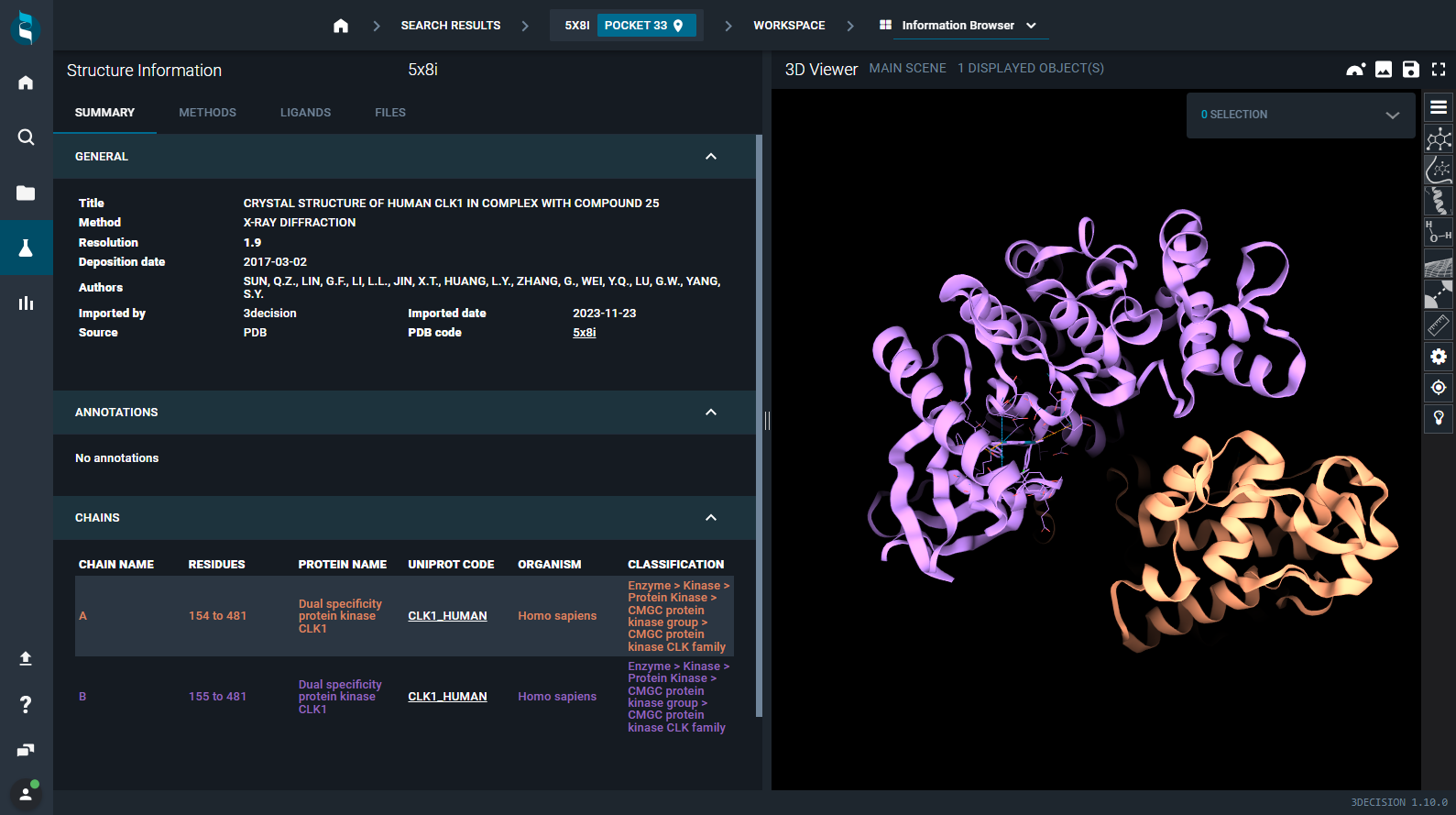
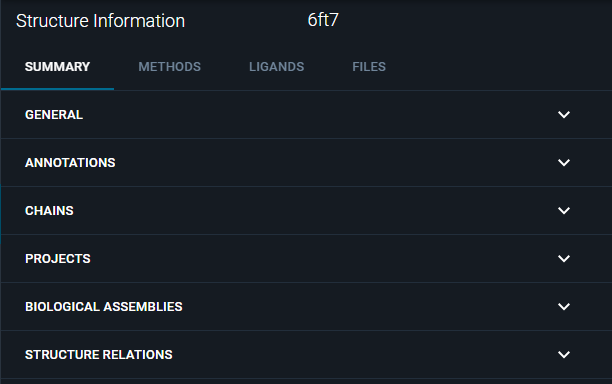
¶ SUMMARY Tab
In this tab, you can retrieve many information about the structure, organized in the subsections:
- GENERAL
- ANNOTATIONS
- CHAINS
- PROJECTS
- BIOLOGICAL ASSEMBLIES
- RELATIONS
In the following sections, each subsection of the SUMMARY tab is described.
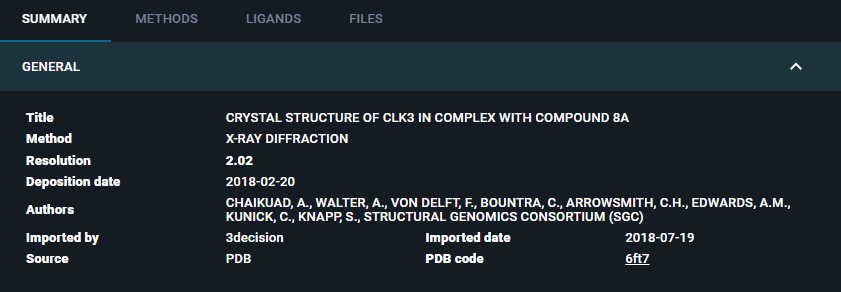
¶ GENERAL
Here you can find the following information:
- Title: title of the structure file
- Label (Optional): if present, label associated with the structure
- Method: the method used to generate the protein structure (X-Ray diffraction, Electron Microscopy, Model, etc.)
- Resolution: the resolution of the structure
- Deposition date: the date when the structure was deposited in the RCSB PDB
- Authors: list of the creators of the structure file
- Imported by: who imported the structure in the 3decision database (for public structures is "3decision"; for proprietary structures is the user who performed the structure import)
- Importe date: the date when the structure was imported in the 3decision database
- Source: source of the data file (PDB, PDB-derived, AlphaFoldDB4, User)
- PDB code/Code: identifier of the file in the 3decision database (for public structures is the PDB code; for proprietary structures is a 6-digits code)
¶ Differences between public and proprietary structures
For publicly available structures:
- Imported by: is
3decision - Source: is
PDBfor data from the RCSB PDB,PDB-derivedfor biological assemblies generated from public structures,AlphaFoldDB4for models from the AlphaFold Database. - Code: is the public 4-digits PDB code (e.g. 6ft7) and have a clickable link to the corresponding RCSB PDB webpage.
For proprietary structures:
- Imported by: is the
User nameof the user who did the structure import, who is also the "Owner" of the structure - Source: is
User - Code: is a unique 6-digits code (e.g. 7k70zp) generated upon registration of the structure in the 3decision database (called
external code).
Reminder: the 6-digits external code is automatically generated for any proprietary structure imported in the 3decision database.
If you wish to add an "Internal Structure ID" to your structure, you should expoit Structure Annotations.
¶ Differences between "Author" and "Owner" of structures
In 3decision, you have an important difference between Authors (corresponding to CREATED_BY property) and Owner (corresponding to IMPORTED_BY property) of the structures:
-
Authors: the author(s) of the structures are the people listed in the PDB header. Therefore, the "Authors" corresponds to the creators of the structure file. In 3decision, "Author" corresponds to the property of the strucure:
CREATED_BY, and it is directly parsed from the PDB file header. Authors are listed in the Information Browser in theAuthorsfield of the STRUCTURE tab, and in theCREATED_BYfield of the Table View. -
Owner: is the user who imported the structure in the database, and is defined in the structure property
IMPORTED_BY. The Owner has full privileges on the structure in the 3decision database and can manage access to the structure.
If no Author is specified in the PDB header, 3decision will consider as the "author" of the structure the user who imported it.
¶ Annotations
You can add annotations to a structure that will be displayed in this section of the Information Browser, organized in a table. These are called "Structure Annotations".
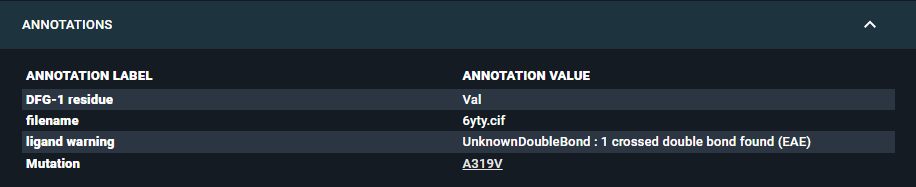
For each structure annotation, you need to define an "Annotation Label" (that is the type of annotation) and an "Annotation Value" (that is the annotation itself). Also, you can add an URL link to the annotation. This can be particularly useful for instance for connecting to internal databases or ELN.
The only structure annotation that can be added upon registration into 3decision is the internal Structure ID, while any other Annotation needs to be added after registration of the structure into the database, using the dedicated API endpoint (see the dedicated API documentation). You can edit structure annotations with the API endpoint PUT /structures/{structure_id}/annotations (instructions here).
Please note that Structure annotations are distinct from the annotations reported in the Annotation Browser (Sequence Annotations) and are not linked to the 3D Viewer.
¶ Chains
The protein chains composing the structure are listed here and color-coded as they appear in the 3D Viewer (with the structure colored by the scheme "by chain").
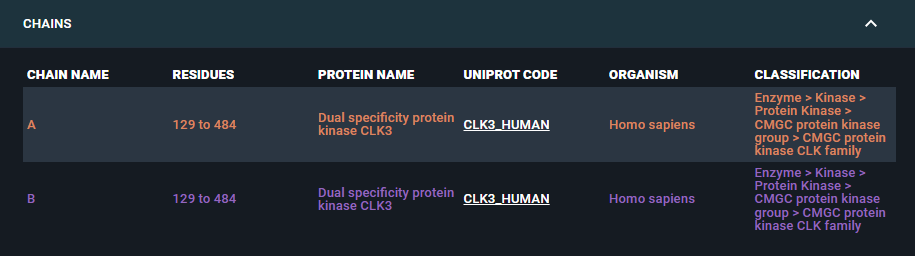
A link to the corresponding UniProt page is provided for each chain.
Chain identification in 3decision is performed through a unique post-registration analysis called sequence mapping. For more details, refer to the dedicated documentation.
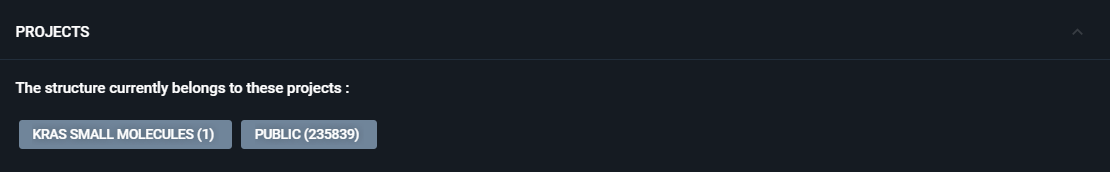
¶ Projects
If the structure is included in one or multiple Projects, they will be displayed in this section.
¶ Biological Assemblies
This section is displayed only for structures associated with at least one biological assembly. All related biological assemblies are listed here. Also, if the structure itself is a bioassembly, the parent structure will be linked in this section.
By clicking on the biological assembly code or the parent structure code, you can quickly visualize it in a new tab or add it to the current workspace.

¶ Structure Relations
If the structure is related to others (for instance through biological assemblies), this will be reported in this section. You can open the related structures in a new tab, or add them to the workspace.
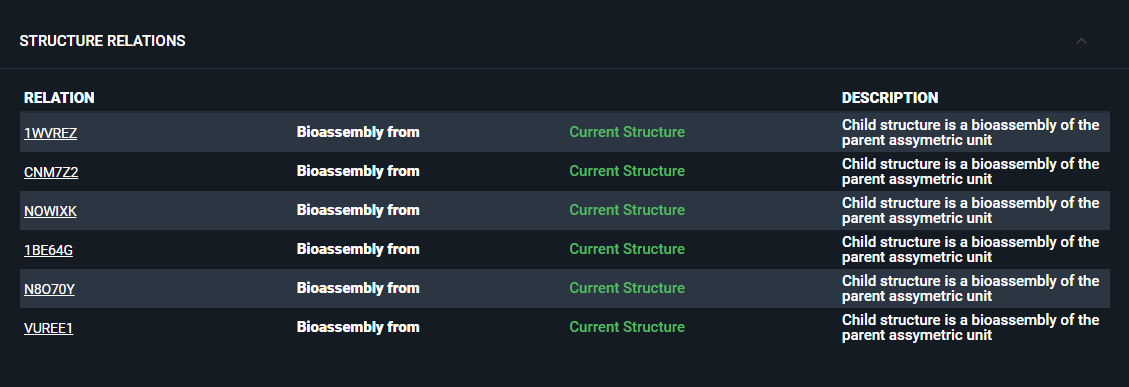
You can add structure relationships when registering a structure in the database. For a step-by-step guide on how to add relationships and additional details, refer to the Structure Registration from UI tutorial.
¶ METHODS Tab
This tab provides a summary of how the structure was produced, with structure parameters categorized and displayed into dedicated sections for a clear overview of the experimental or modeling details. Both PDBx/mmCIF and ModelCIF categories are parsed.
When importing a structure into 3decision, users can provide either a PDB file or an mmCIF file as input. Regardless of the input format, 3decision always generates an enriched mmCIF file during the import process. This enriched file contains standardized and augmented structural metadata derived from the original input. The structure parameters displayed in the Information Browser are extracted by parsing this enriched mmCIF file.
If you want specific structure parameters from your original structure file to appear in the Information Browser, it’s essential to understand how the user interface maps those values. The sections below detail the exact correspondence between mmCIF categories/data items and their display labels in the UI. Where relevant, the equivalent PDB record keywords are also provided for structures supplied in PDB format:
Structure Parameter Label - PDB/mmCIF Field Correspondence
¶ 1. Experiment Summary
Basic information about the experimental method used to determine the structure.
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Experimental Method | _exptl.method |
EXPDTA: Experimental_Technique |
| Reconstruction Method | _em_experiment.reconstruction_method |
REMARK 245: RECONSTRUCTION METHOD |
| Aggregation State | _em_experiment.aggregation_state |
REMARK 245: SAMPLE TYPE |
¶ 2. Cell
Unit cell parameters describing the crystal lattice geometry.
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| a, b, c | _cell.length_a, _cell.length_b, _cell.length_c |
CRYST1: a, b, c |
| α, β, γ | _cell.angle_alpha, _cell.angle_beta, _cell.angle_gamma |
CRYST1: Alpha, Beta, Gamma |
| Space Group | _symmetry.space_group_name_H-M |
CRYST1: Space_Group |
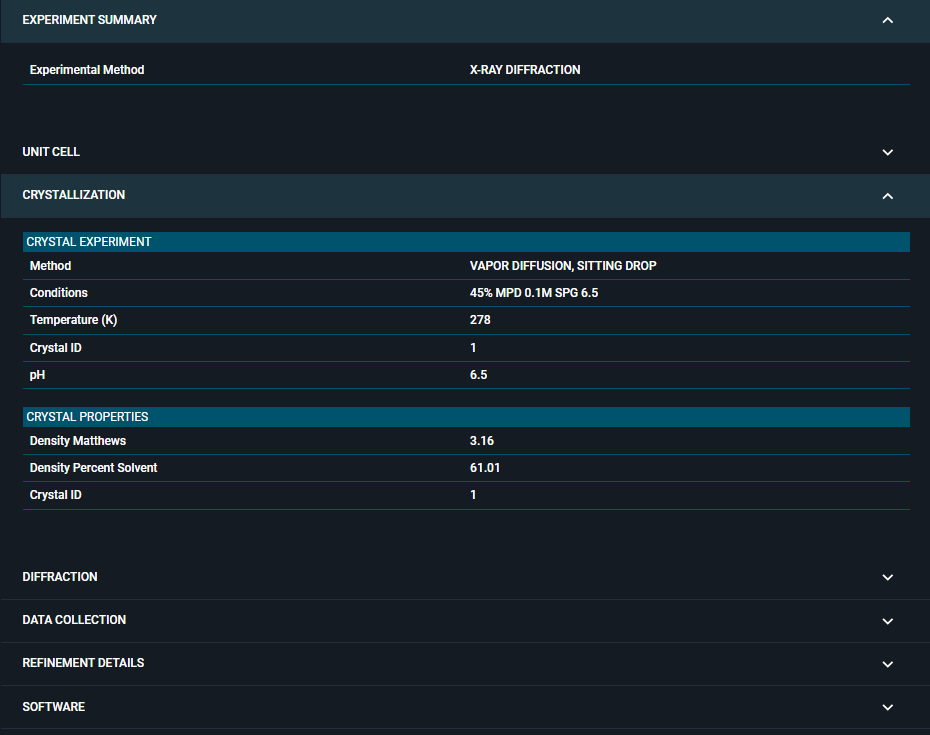
¶ 3. Crystallization
Information about crystal growth conditions.
¶ Crystal Experiment
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Crystal ID | _exptl_crystal_grow.crystal_id |
N/A |
| Method | _exptl_crystal_grow.method |
N/A |
| pH | _exptl_crystal_grow.pH |
REMARK 200/280: PH |
| Pressure | _exptl_crystal_grow.pressure |
N/A |
| Temperature (K) | _exptl_crystal_grow.temp |
N/A |
| Conditions | _exptl_crystal_grow.pdbx_details |
REMARK 280: CRYSTALLIZATION CONDITIONS |
¶ Crystal Properties
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Colour | _exptl_crystal.colour |
N/A |
| Density Matthews | _exptl_crystal.density_Matthews |
REMARK 280: MATTHEWS COEFFICIENT |
| Density Percent Solvent | _exptl_crystal.density_percent_sol |
REMARK 280: SOLVENT CONTENT |
| Description | _exptl_crystal.description |
REMARK 200: REMARK |
¶ 4. Diffraction
Information about X-ray diffraction data collection.
¶ Diffraction Experiment
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Scattering Type | _diffrn_radiation.pdbx_scattering_type |
N/A |
| Ambient Temperature (K) | _diffrn.ambient_temp |
REMARK 200: TEMPERATURE |
| Detector | _diffrn_detector.detector |
REMARK 200: DETECTOR TYPE |
| Detector Type | _diffrn_detector.type |
REMARK 200: DETECTOR MANUFACTURER |
| Collection Date | _diffrn_detector.pdbx_collection_date |
REMARK 200: DATE OF DATA COLLECTION |
| Monochromatic (M) or Laue (L) | _diffrn_radiation.pdbx_monochromatic_or_laue_m_l |
REMARK 200: MONOCHROMATIC OR LAUE |
| Diffraction Protocol | _diffrn_radiation.pdbx_diffrn_protocol |
REMARK 200: DIFFRACTION PROTOCOL |
¶ Radiation Source
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Source | _diffrn_source.source |
REMARK 200 SYNCHROTRON (Y/N) |
| Source Type | _diffrn_source.type |
REMARK 200: X-RAY GENERATOR MODEL |
| Wavelength | _diffrn_source.pdbx_wavelength |
REMARK 200: WAVELENGTH OR RANGE |
| Wavelength List | _diffrn_source.pdbx_wavelength_list |
N/A |
| Synchrotron Site | _diffrn_source.pdbx_synchrotron_site |
REMARK 200 RADIATION SOURCE |
| Synchrotron Beamline | _diffrn_source.pdbx_synchrotron_beamline |
REMARK 200: BEAMLINE |
¶ 5. Data Collection
Statistics about the diffraction data quality.
¶ Overall Statistics
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Resolution (High) | _reflns.d_resolution_high |
REMARK 200: RESOLUTION RANGE HIGH |
| Resolution (Low) | _reflns.d_resolution_low |
REMARK 200: RESOLUTION RANGE LOW |
| Percentage Possible (Observed) | _reflns.percent_possible_obs |
REMARK 200: COMPLETENESS FOR RANGE |
| R Merge I (Observed) | _reflns.pdbx_Rmerge_I_obs |
REMARK 200: R MERGE (I) |
| CC (Half) | _reflns.pdbx_CC_half |
N/A |
| Net I over Average Sigma I | _reflns.pdbx_netI_over_sigmaI |
REMARK 200: <I/SIGMA(I)> FOR THE DATA SET |
| Redundancy | _reflns.pdbx_redundancy |
REMARK 200: DATA REDUNDANCY |
| Number Reflections (All) | _reflns.number_all |
N/A |
| Number Reflections (Observed) | _reflns.number_obs |
REMARK 200: NUMBER OF UNIQUE REFLECTIONS |
| Observed Criterion Sigma (F) | _reflns.observed_criterion_sigma_F |
N/A |
| Observed Criterion Sigma (I) | _reflns.observed_criterion_sigma_I |
REMARK 200: REJECTION CRITERIA (SIGMA(I)) |
| B (Isotropic) From Wilson Plot | _reflns.B_iso_Wilson_estimate |
REMARK 3: FROM WILSON PLOT |
¶ Highest Resolution Shell
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Resolution (High) | _reflns_shell.d_res_high |
REMARK 200: HIGHEST RESOLUTION SHELL, RANGE HIGH |
| Resolution (Low) | _reflns_shell.d_res_low |
REMARK 200: HIGHEST RESOLUTION SHELL, RANGE LOW |
| CC half | _reflns_shell.pdbx_CC_half |
N/A |
| Percent Possible (All) | _reflns_shell.percent_possible_all |
REMARK 200: COMPLETENESS FOR SHELL |
| Percent Possible (Observed) | _reflns_shell.percent_possible_obs |
N/A |
| R Merge I (Observed) | _reflns_shell.Rmerge_I_obs |
REMARK 200: R MERGE FOR SHELL (I) |
| Mean I over Sigma (Observed) | _reflns_shell.meanI_over_sigI_obs |
REMARK 200: <I/SIGMA(I)> FOR SHELL |
| Redundancy | _reflns_shell.pdbx_redundancy |
REMARK 200: DATA REDUNDANCY IN SHELL |
| Number Unique Reflections (Observed) | _reflns_shell.number_unique_obs |
N/A |
| Number Unique Reflections (All) | _reflns_shell.number_unique_all |
N/A |
¶ 6. Electron Microscopy
Parameters specific to cryo-EM structure determination.
¶ Sample
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Assembly Name | _em_entity_assembly.name |
REMARK 245: NAME OF SAMPLE |
| Assembly Type | _em_entity_assembly.type |
N/A |
| Assembly Source | _em_entity_assembly.source |
N/A |
¶ Specimen Preparation
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Specimen Concentration | _em_specimen.concentration |
REMARK 245: SAMPLE CONCENTRATION |
| pH | _em_buffer.pH |
REMARK 245: PH |
| Instrument | _em_vitrification.instrument |
N/A |
| Vitrification Cryogen | _em_vitrification.cryogen_name |
N/A |
| Vitrification Details | _em_vitrification.details |
REMARK 245: SAMPLE VITRIFICATION DETAILS |
¶ Imaging
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Microscope Model | _em_imaging.microscope_model |
REMARK 245: MICROSCOPE MODEL |
| Illumination Mode | _em_imaging.illumination_mode |
REMARK 245: ILLUMINATION MODE |
| Imaging Mode | _em_imaging.mode |
REMARK 245: IMAGING MODE |
| Electron Source | _em_imaging.electron_source |
REMARK 245: SOURCE |
| Accelerating Voltage (kV) | _em_imaging.accelerating_voltage |
REMARK 245: ACCELERATION VOLTAGE |
| Specimen Holder Model | _em_imaging.specimen_holder_model |
N/A |
| Cooling Holder Cryogen | _em_imaging.cryogen |
N/A |
¶ Specialist Optics
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Energy Filter Name | _em_imaging_optics.energyfilter_name |
N/A |
| Energy Filter Slit Width (eV) | _em_imaging_optics.energyfilter_slit_width |
N/A |
¶ Image Recording
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Film or Detector Model | _em_image_recording.film_or_detector_model |
REMARK 245: DETECTOR TYPE |
| Average Electron Dose per Image (e/Ų) | _em_image_recording.avg_electron_dose_per_image |
REMARK 245: ELECTRON DOSE |
¶ Final Reconstruction
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| 3D Resolution | _em_3d_reconstruction.resolution |
REMARK 3: EFFECTIVE RESOLUTION (ANGSTROMS) |
| 3D Resolution Method | _em_3d_reconstruction.resolution_method |
N/A |
| Number of Particles | _em_3d_reconstruction.num_particles |
REMARK 3: NUMBER OF PARTICLES |
| 3D Symmetry Type | _em_3d_reconstruction.symmetry_type |
REMARK 245: PARTICLE TYPE |
¶ Applied Symmetry
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Point Symmetry | _em_single_particle_entity.point_symmetry |
N/A |
| Axial Symmetry | _em_helical_entity.axial_symmetry |
N/A |
¶ 7. NMR Experiment
Parameters for solution NMR structure determination.
¶ Sample
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Solution ID | _pdbx_nmr_sample_details.solution_id |
N/A |
| Contents | _pdbx_nmr_sample_details.contents |
REMARK 210: SAMPLE CONTENTS |
¶ Conditions
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Condition ID | _pdbx_nmr_exptl_sample_conditions.conditions_id |
N/A |
| Ionic Strength | _pdbx_nmr_exptl_sample_conditions.ionic_strength |
REMARK 210: IONIC STRENGTH |
| pH | _pdbx_nmr_exptl_sample_conditions.pH |
REMARK 210: PH |
| Pressure | _pdbx_nmr_exptl_sample_conditions.pressure |
REMARK 210: PRESSURE |
| Pressure Units | _pdbx_nmr_exptl_sample_conditions.pressure_units |
N/A |
| Temperature | _pdbx_nmr_exptl_sample_conditions.temperature |
REMARK 210: TEMPERATURE |
| Temperature Units | _pdbx_nmr_exptl_sample_conditions.temperature_units |
N/A |
¶ Experiment
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Experiment ID | _pdbx_nmr_exptl.experiment_id |
N/A |
| Conditions ID | _pdbx_nmr_exptl.conditions_id |
N/A |
| Solution ID | _pdbx_nmr_exptl.solution_id |
N/A |
| Type | _pdbx_nmr_exptl.type |
REMARK 210: NMR EXPERIMENTS CONDUCTED |
| Sample State | _pdbx_nmr_exptl.sample_state |
N/A |
¶ Spectrometer
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Manufacturer | _pdbx_nmr_spectrometer.manufacturer |
REMARK 210: SPECTROMETER MANUFACTURER |
| Model | _pdbx_nmr_spectrometer.model |
REMARK 210: SPECTROMETER MODEL |
| Field Strength | _pdbx_nmr_spectrometer.field_strength |
REMARK 210: SPECTROMETER FIELD STRENGTH |
¶ Refinement
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Method | _pdbx_nmr_refine.method |
REMARK 210: METHOD USED |
| Details | _pdbx_nmr_refine.details |
N/A |
¶ Ensemble
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Conformer Selection Criteria | _pdbx_nmr_ensemble.conformer_selection_criteria |
REMARK 210: CONFORMERS, SELECTION CRITERIA |
| Total Conformers Submitted | _pdbx_nmr_ensemble.conformers_submitted_total_number |
REMARK 210: CONFORMERS, NUMBER SUBMITTED |
| Total Conformers Calculated | _pdbx_nmr_ensemble.conformers_calculated_total_number |
REMARK 210: CONFORMERS, NUMBER CALCULATED |
| Representative Model | _pdbx_nmr_representative.conformer_id |
REMARK 210: BEST REPRESENTATIVE CONFORMER |
| Representative Selection Criteria | _pdbx_nmr_representative.selection_criteria |
N/A |
¶ 8. Refinement Details
Information about structure refinement against experimental data.
¶ Statistics
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Refinement ID | _refine.pdbx_refine_id |
N/A |
| Structure Solution Method | _refine.pdbx_method_to_determine_struct |
REMARK 200: METHOD USED TO DETERMINE THE STRUCTURE |
| Cross Validation Method | _refine.pdbx_ls_cross_valid_method |
REMARK 3: CROSS-VALIDATION METHOD |
| Starting Model | _refine.pdbx_starting_model |
REMARK 200: STARTING MODEL |
| Resolution (High) | _refine.ls_d_res_high |
REMARK 3: RESOLUTION RANGE HIGH |
| Resolution (Low) | _refine.ls_d_res_low |
REMARK 3: RESOLUTION RANGE LOW |
| Cut-off Sigma (F) | _refine.pdbx_ls_sigma_F |
REMARK 3: DATA CUTOFF (SIGMA(F)) |
| Number Reflections (Observed) | _refine.ls_number_reflns_obs |
REMARK 3: NUMBER OF REFLECTIONS |
| Number Reflections (R-Free) | _refine.ls_number_reflns_R_free |
REMARK 3: FREE R VALUE TEST SET COUNT |
| Percent Reflections (Observed) | _refine.ls_percent_reflns_obs |
REMARK 3: COMPLETENESS (WORKING+TEST) |
| R-Factor (Observed) | _refine.ls_R_factor_obs |
REMARK 3: R VALUE (WORKING + TEST SET) |
| R-Work | _refine.ls_R_factor_R_work |
REMARK 3: R VALUE (WORKING SET) |
| R-Free | _refine.ls_R_factor_R_free |
REMARK 3: FREE R VALUE |
| R-Free Selection Details | _refine.pdbx_R_Free_selection_details |
REMARK 3: FREE R VALUE TEST SET SELECTION |
| Mean Isotropic B | _refine.B_iso_mean |
REMARK 3: MEAN B VALUE (OVERALL) |
¶ Temperature Factor Modeling
Anisotropic B-factor tensor components (if refined anisotropically):
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Aniso B[1][1] | _refine.aniso_B[1][1] |
REMARK 3: B11 |
| Aniso B[1][2] | _refine.aniso_B[1][2] |
REMARK 3: B12 |
| Aniso B[1][3] | _refine.aniso_B[1][3] |
REMARK 3: B13 |
| Aniso B[2][1] | _refine.aniso_B[2][1] |
REMARK 3: B21 |
| Aniso B[2][2] | _refine.aniso_B[2][2] |
REMARK 3: B22 |
| Aniso B[2][3] | _refine.aniso_B[2][3] |
REMARK 3: B23 |
| Aniso B[3][1] | _refine.aniso_B[3][1] |
REMARK 3: B31 |
| Aniso B[3][2] | _refine.aniso_B[3][2] |
REMARK 3: B32 |
| Aniso B[3][3] | _refine.aniso_B[3][3] |
REMARK 3: B33 |
¶ Non-Hydrogen Atoms Used in Refinement
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Number of Protein Atoms | _refine_hist.pdbx_number_atoms_protein |
REMARK 3: PROTEIN ATOMS |
| Number of Nucleic Acid Atoms | _refine_hist.pdbx_number_atoms_nucleic_acid |
REMARK 3: NUCLEIC ACID ATOMS |
| Number of Solvent Atoms | _refine_hist.number_atoms_solvent |
REMARK 3: SOLVENT ATOMS |
| Number of Heterogen Atoms | _refine_hist.pdbx_number_atoms_ligand |
REMARK 3: HETEROGEN ATOMS |
¶ 9. Software
Programs used during structure determination.
¶ Software (X-ray/General)
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Ordinal | _software.pdbx_ordinal |
N/A |
| Function | _software.classification |
N/A |
| Program | _software.name |
REMARK 3: PROGRAM |
| Version | _software.version |
N/A |
¶ EM Software
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Category | _em_software.category |
N/A |
| Name | _em_software.name |
REMARK 3: SOFTWARE PACKAGES |
| Version | _em_software.version |
N/A |
¶ NMR Software
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Function | _pdbx_nmr_software.classification |
N/A |
| Program | _pdbx_nmr_software.name |
REMARK 210: SOFTWARE USED |
| Version | _pdbx_nmr_software.version |
N/A |
¶ Model Software (AlphaFold/Computed Models)
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Group ID | _ma_software_group.group_id |
N/A |
| Software ID | _ma_software_group.software_id |
N/A |
| Parameter Group ID | _ma_software_group.parameter_group_id |
N/A |
¶ Model Software Parameters
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Group ID | _ma_software_parameter.group_id |
N/A |
| Name | _ma_software_parameter.name |
N/A |
| Value | _ma_software_parameter.value |
N/A |
| Description | _ma_software_parameter.description |
N/A |
¶ Model Software Protocols
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Step ID | _ma_protocol_step.step_id |
N/A |
| Step Name | _ma_protocol_step.step_name |
N/A |
| Method Type | _ma_protocol_step.method_type |
N/A |
| Software Group ID | _ma_protocol_step.software_group_id |
N/A |
¶ 10. TLS Groups
Translation/Libration/Screw (TLS) refinement parameters for modeling anisotropic motion.
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Details | _pdbx_refine_tls.details |
N/A |
| Method | _pdbx_refine_tls.method |
N/A |
| Origin X | _pdbx_refine_tls.origin_x |
REMARK 3: ORIGIN FOR THE GROUP (A) |
| Origin Y | _pdbx_refine_tls.origin_y |
REMARK 3: ORIGIN FOR THE GROUP (A) |
| Origin Z | _pdbx_refine_tls.origin_z |
REMARK 3: ORIGIN FOR THE GROUP (A) |
| T[1][1] through T[3][3] | _pdbx_refine_tls.T[i][j] |
REMARK 3: T TENSOR |
| L[1][1] through L[3][3] | _pdbx_refine_tls.L[i][j] |
REMARK 3: L TENSOR |
| S[1][1] through S[3][3] | _pdbx_refine_tls.S[i][j] |
REMARK 3: S TENSOR |
¶ 11. Model References
Information for computed/predicted models (e.g., AlphaFold).
¶ Data Used in Modeling
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Model ID | _ma_data.id |
N/A |
| Model Name | _ma_data.name |
N/A |
| Model Type | _ma_data.content_type |
N/A |
¶ List of Models
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Model ID | _ma_model_list.model_id |
N/A |
| Model Group ID | _ma_model_list.model_group_id |
N/A |
| Model Name | _ma_model_list.model_name |
N/A |
| Model Group Name | _ma_model_list.model_group_name |
N/A |
| Assembly ID | _ma_model_list.assembly_id |
N/A |
| Data ID | _ma_model_list.data_id |
N/A |
| Model Type | _ma_model_list.model_type |
N/A |
¶ Target References
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Target Entity ID | _ma_target_ref_db_details.target_entity_id |
N/A |
| Database Name | _ma_target_ref_db_details.db_name |
N/A |
| Database Code | _ma_target_ref_db_details.db_code |
N/A |
| Database Accession | _ma_target_ref_db_details.db_accession |
N/A |
| Sequence Database Isoform | _ma_target_ref_db_details.seq_db_isoform |
N/A |
| Sequence Database Align Begin | _ma_target_ref_db_details.seq_db_align_begin |
N/A |
| Sequence Database Align End | _ma_target_ref_db_details.seq_db_align_end |
N/A |
¶ Template References
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Ordinal ID | _ma_template_details.ordinal_id |
N/A |
| Template ID | _ma_template_details.template_id |
N/A |
| Template Origin | _ma_template_details.template_origin |
N/A |
| Template Entity Type | _ma_template_details.template_entity_type |
N/A |
¶ Template DB References
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Template ID | _ma_template_ref_db_details.template_id |
N/A |
| Database Name | _ma_template_ref_db_details.db_name |
N/A |
| Database Accession Code | _ma_template_ref_db_details.db_accession_code |
N/A |
¶ Associated Files
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Entry ID | _ma_entry_associated_files.entry_id |
N/A |
| File Content | _ma_entry_associated_files.file_content |
N/A |
| Details | _ma_entry_associated_files.details |
N/A |
| File Type | _ma_entry_associated_files.file_type |
N/A |
| File Format | _ma_entry_associated_files.file_format |
N/A |
| File URL | _ma_entry_associated_files.file_url |
N/A |
¶ QA Metrics
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Metric ID | _ma_qa_metric.id |
N/A |
| Metric Name | _ma_qa_metric.name |
N/A |
| Metric Type | _ma_qa_metric.type |
N/A |
| Metric Mode | _ma_qa_metric.mode |
N/A |
| Software Group ID | _ma_qa_metric.software_group_id |
N/A |
¶ Global QA Metrics
| Field | mmCIF Data Item | PDB Field Name |
|---|---|---|
| Model ID | _ma_qa_metric_global.model_id |
N/A |
| Metric ID | _ma_qa_metric_global.metric_id |
N/A |
| Metric Value | _ma_qa_metric_global.metric_value |
N/A |
The displayed sections vary based on the method used for structure determination:
- X-Ray diffraction structures include crystallization conditions and diffraction data statistics
- Cryo-EM structures contain details on specimen preparation, imaging, and reconstruction
- NMR structures show experimental conditions, spectrometer details, and ensemble information
- Computed models (e.g., AlphaFold) display information on the modeling tool, protocols, and quality metrics
¶ LIGANDS Tab
In this tab, all the ligands in the structure are listed in ligand cards. LigandID annotations can be found indicated in the ligand card.
¶ FILES Tab
The Files tabs contains all the available structure files and any associated files to structure (e.g., electron density maps).
The tab is divided into three sections (details on each section provided below):
- 3decision structure files
- Original structure file
- Associated files
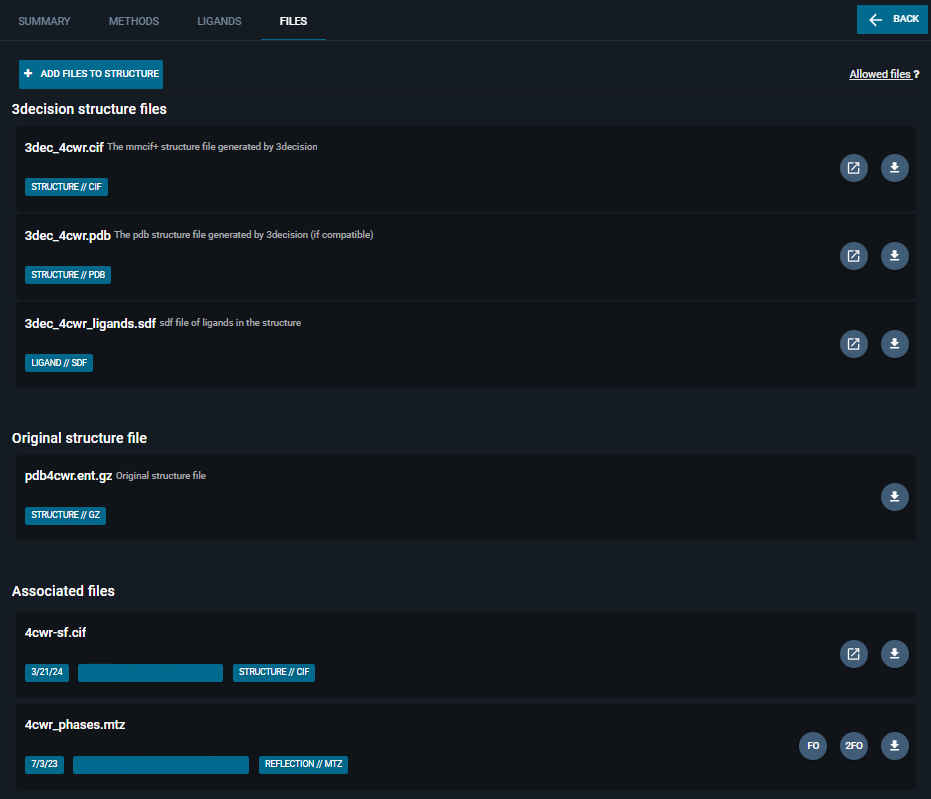
¶ 3decision structure files
For each structure included in the 3decision database, the following files can be downloaded:
- an enriched .cif file (called mmCIF+): containing all the relevant structure information in a standardized manner. Some unique fields can be found in this structure file, such as:
_entity_3decor_entity_ccg: a list of the recognized non proteic entities, their SMILES, InChI and aisligandflag_3dec_summaryor_ccg_summary: a list of the basic structure info such as external code, label and experimental method.
- a .PDB structure file (if the structure is compatible with the PDB standards);
- The original structure file: the file firstly uploaded (either PDB or mmCIF);
- an .sdf file of all ligands in the structure, if any ligand is present.
¶ Original Structure File
You can find here the original structure file that was provided upon structure registration, without any 3decision modification.
¶ Associated files
To each structure in the database, you can attach additional associated files:
- from the UI, using the blue button on the top-left "Add files to structure";
- from the API, with the endpoint
POST /structures/file/{structure_id}(step-by-step guide here)
Compatible files formats are:
.brix .ccp4 .cif .cns .csv .cube .dcd .doc .docx .dsn6 .dx .dxbin .ent .fasta .gro .jpeg .jpg .log .map .map.gz .mmcif .mmtf .mol .mol2 .mrc .mrc.gz .mtz .ncdf .nctraj .pdb .pdf .png .ppt .pptx .pqr .prmtop .psf .rtf .sd .sdf .top .trr .txt .xls .xlsx .xplor .xtc .zip
For electron density maps and mtz files, you have dedicated buttons on the file card for visualizing the electron density map in the 3DViewer.