This section of the User Guide describes the Pocket Similarity Search algorithm used in 3decision up to version 2.2.
For version 2.3 and later, refer to this section of the documentation, that describes the revamped and enhanced Pocket Search algorithm.
¶ Concepts
One of the most distinctive features of 3decision is its nearly complete indexing of known and putative binding sites. 3decision currently contains a functionality allowing you to search for similar protein binding sites or subparts of these. This allows to quickly find structurally similar local environments and get information on possible small molecule / peptide fragments that might bind to similar locations. It can also indicate potential off-targets for example that one needs to monitor during compound design cycles within a project.
¶ Subpockets vs Pockets
Nearly 100% of the literature focuses on comparing full binding sites (so a whole pocket). On the contrary, the algorithm and data-structures integrated in 3decision allow to run sub-pocket comparisons, i.e. comparisons of bits and pieces of pockets. This is algorithmically more complex and no evaluation or benchmark exists yet for such use-cases in literature. You can however run full binding site comparisons as well.
¶ Comparison and benchmark metrics
Most of the methods and benchmarks are set up in a way that one has to run one by one comparison of binding sites. For example, is my ATP binding site of 4CWR similar to my ATP binding site of 4GFO (both, different proteins)? In general you would get back a comparison score (similarity) and then you can rank all comparisons and get performance measures like TPR vs FPR, precision-recall curves, F1 measures (if you want to assess scores).
This is not how 3decision's pocket search is set up. 3decision's pocket comparison is set up from a database search / retrieval perspective, so given a query pocket or subpocket, it will allow you to retrieve all pockets and subpockets that match that query given a set of parameters for the search.
¶ How does the pocket comparison algorithm work ?
¶ Making a pocket searchable
- Identify binding sites using fpocket (knowing where the ligand is, and without any ligand knowledge)
- For each binding site (sufficiently big, i.e. containing at least 30 alpha spheres or any ligand), characterize pocket features
- For each binding site lining residue, check what physicochemical features are exposed to the surface of the binding site (contribution to potential interactants)
Schematic representation of the feature detection and definition process on a tyrosine. Physico-chemical features on residues lining the binding site are identified and it is assessed if they contribute to the accessible surface area of the pocket. The Cα position is tracked and the presence and absence of a feature is tracked in a 6 bit binary vector, each bit representing one possible feature.
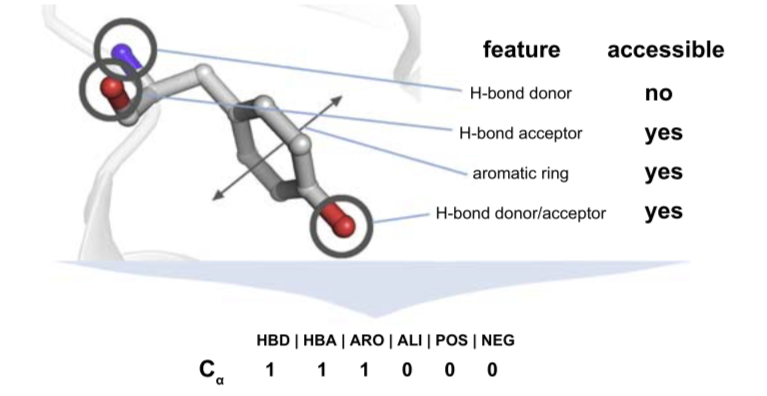
- Encode these features as fingerprint (1 if present, 0 if not) for hydrophobic, negatively charged, positively charged, HBD/A, aromatics...
- Register Cα position of the residue together with the fingerprint to the database attached to the pocket
- Register pocket feature pairs
- For each pair of features in a binding site (in a given set of distance range) in a canonical order: distance, but also relative angle in local amino acid space is calculated for the vector defining a feature pair from residue 1 (feature), to residue 2 (another feature)
¶ Searching for a pocket
Searching for a binding site starts with a set of residues (features) defined by the user on a particular binding site of a particular structure. The main search has now been ported nearly entirely to an SQL query. It roughly goes through the following steps:
- Get all feature pairs involved in the selected residues or including at least one of the residues selected (reference feature pairs)
- Find all other sets feature pairs that do not deviate too much from the angles and distances of the reference feature pairs (sort of a maximum clique detection thingy thing)
- Postprocess results to determine whether identified hits are topologically relevant (checking consistency of distances between matched feature pair vectors between hit and reference pairs).
¶ Pocket Similarity Search
¶ Access
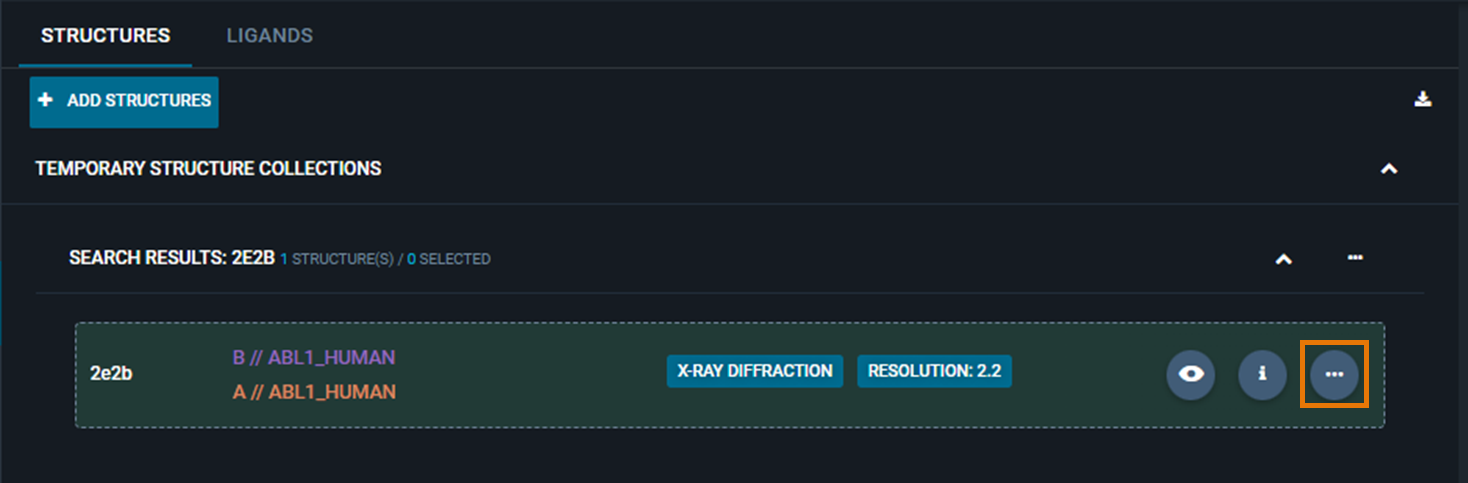
There are two ways to access Pocket Similarity Search in 3decision. To run a pocket comparison, your structure of interest has to be loaded in the 3D viewer first.
- Click on the three-dot button (Structure Actions) on the same line of your structure, then click on "Find similar pockets"
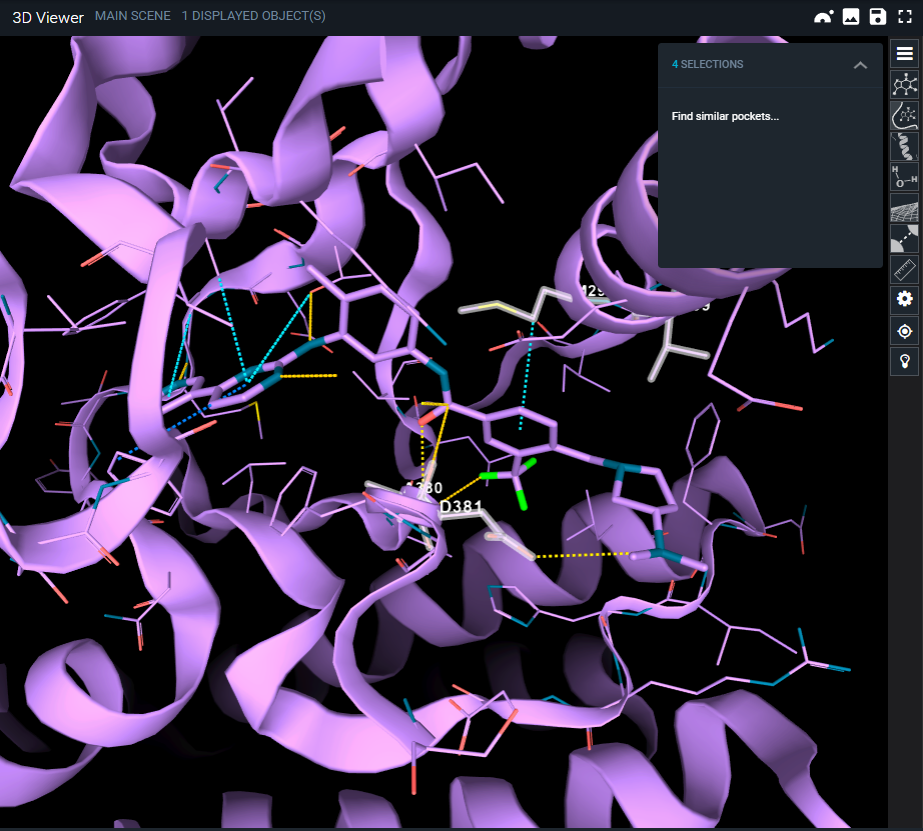
2. On top of the 3D viewer, near SELECTION, you can click on the little arrow. It will open a menu with "Find similar pockets..."
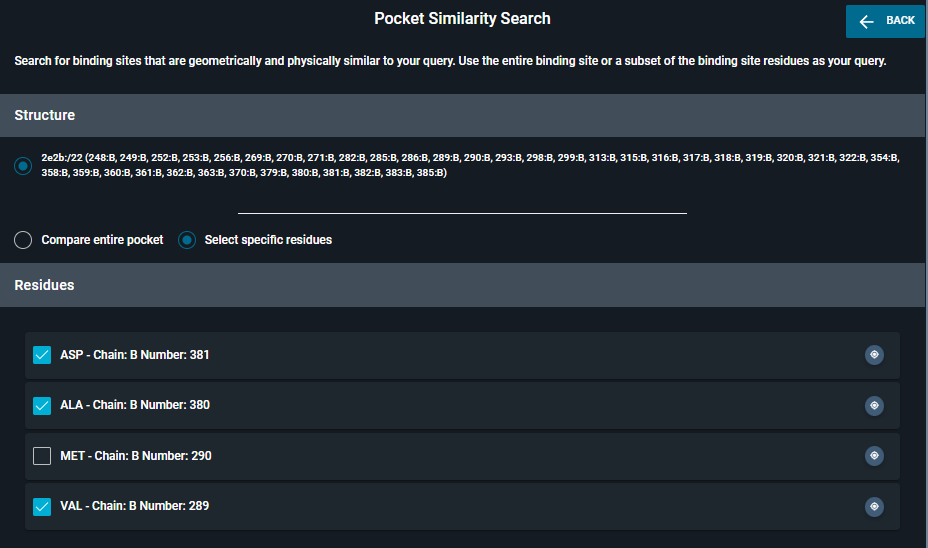
Once the panel open, you have the choice between
- Compare entire pocket
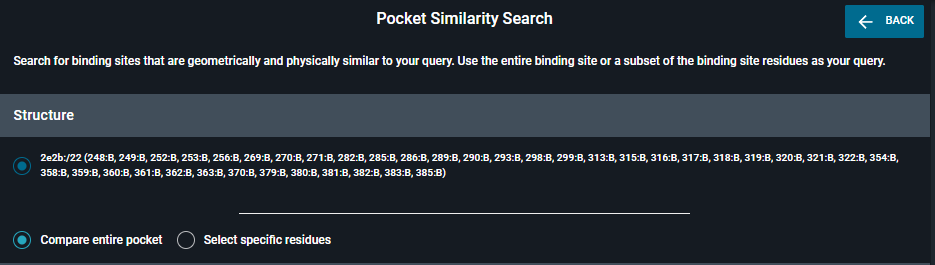
- Select specific residues (to do subpocket search)
You can click directly in the 3D viewer, or use the shortcut Alt+left-click on a ligand atom to select residues. They will appear in the section Residues.
Once in the list, you can deselect one residue by clicking on the blue square.
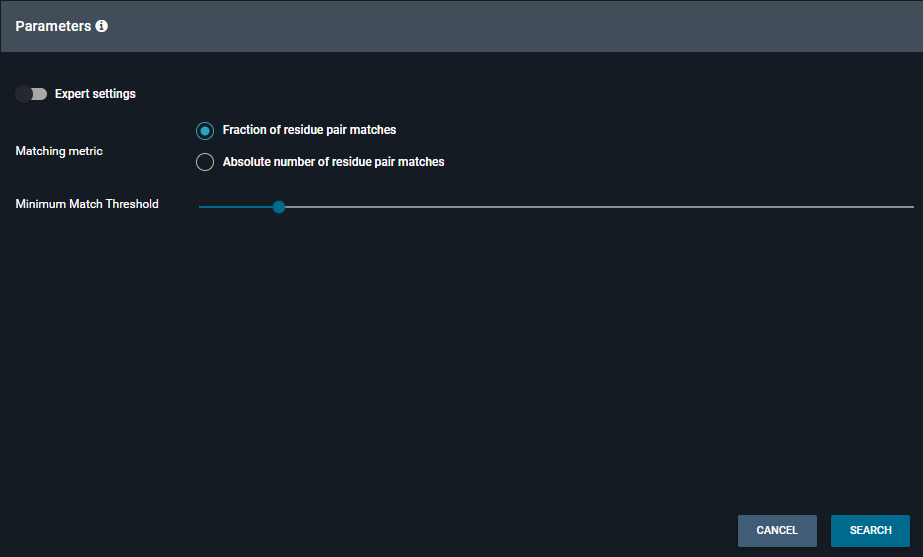
¶ Parameters
To define the parameters, two modes are available.
- In normal mode, you can choose the Matching metric :
- Fraction of residue pair matches (Minimum Match Treshold from 0 to 1)
- Absolute number of residue pair matches (Minimum Match Treshold from 3 to 50)
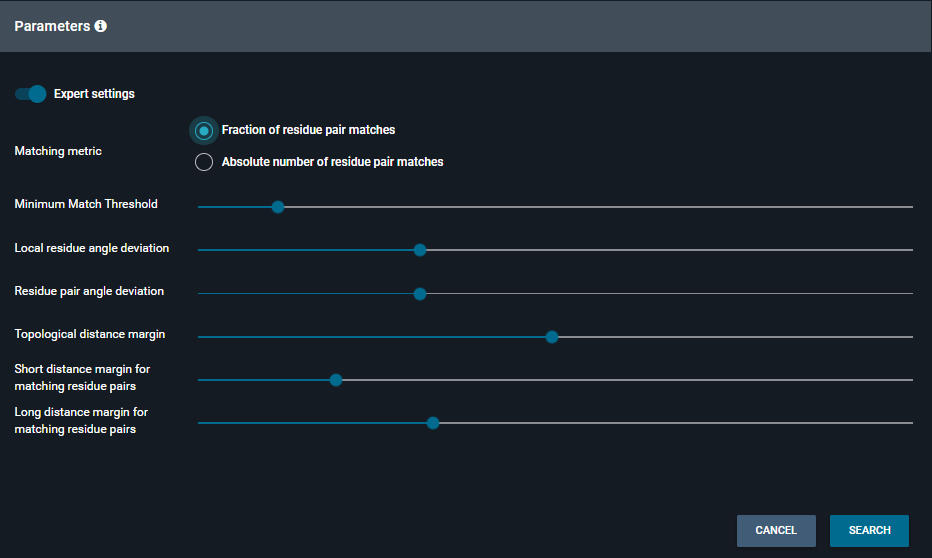
- In expert mode, there are more parameters you can play with:
- Minimum Match Threshold: how many features pairs of the reference pocket need to be matched for a hit pocket to be valid and returned (fraction or absolute, depending on the Matching metric selected)
- Local residue angle deviation: allowed deviation for local residue pair angles (in radians) of query versus hit residue pair. The higher, the fuzzier the search.
- Residue pair angle deviation: allowed deviation of angles (in radians) between two matching residue pairs between two binding sites
- Topological distance margin: maximum allowed distance in Angströms between two residue matched pairs (center of vector distances are compared)
- Short distance margin for matching residue pairs: deviation of the short distance allowed to identify a match between feature pairs
- Long distance margin for matching residue pairs: deviation of the long distance allowed to identify a match between feature pairs
¶ FAQ
¶ I only get hits on the same protein as the query structure:
Either there is no further information to be found, or your search parameters are too stringent.
Try to loosen the local angle threshold or switch to number of residue matches > 3 or 4.
¶ My search runs forever without returning a result:
This should not happen, please reach out to 3decision-support@discngine.com with the parameters of your search so we can fix this.
¶ I have too many times the same biomolecule returned, can't I filter them out?
Not yet, but we are planning on integrating a filter so you can focus only on different biomolecular structure or those that are different to your query structure.
¶ Benchmarking
¶ Benchmark sets and first results
In order to show that our integration of subpocket searches works in the context of general pocket comparison, one obvious thing to test is whether more or less the same binding site on the same protein, but in different structures can be found if there are conformational changes or not.
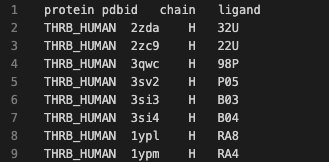
One could easily prepare a set of structures of good quality on the same biomolecule sequence, encompassing the same domain and binding site overall. This has been done already to a small extent by the paper cited above, where Dataset 1 (https://journals.plos.org/ploscompbiol/article/file?type=supplementary&id=10.1371/journal.pcbi.1006483.s010) is a list of PDB structures, the chain and ligand code to identify the binding site they were working on. The computer digestable file looks a bit like this:
The following tests aim to show you the impact of some of the parameters exposed in the 3decision front-end to the final results. These are preliminary benchmark results and performance & parameter sets will likely evolve in future releases.
¶ First test
The first test we ran is for each pocket in the dataset. :
- Retrieve all residues of that binding site using the ligand and chain definition from an explicit pocket
- Run a subpocket search with all residues and default parameters
- Analyze for each search how many hits from the same protein have been identified (tpr) or have not been found.
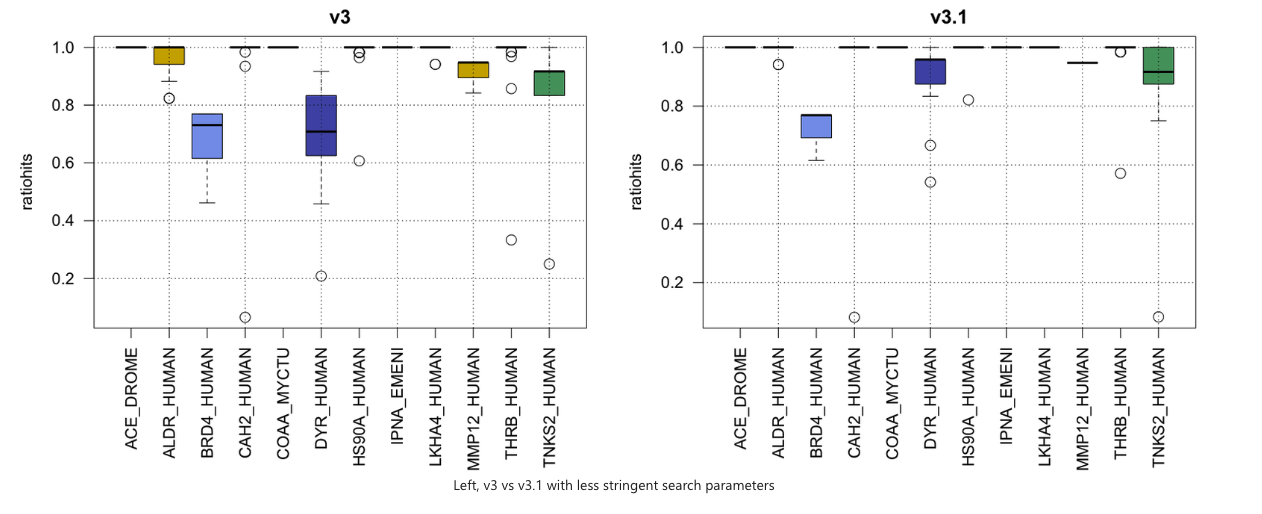
This was done with two different parameter sets. On the left hand side one more stringent one, on the right hand side a more relaxed one.
What we want to achieve here is, to have every box at a 100% true positive rate ideally. So for instance for HSP90 (HS90_HUMAN), if you take whatever binding site from whatever structure you are sure to retrieve nearly all of the others in the database.
The parameter set on the right as an example was:
- Maximum angular deviation: 0.15
- Filtering criteria: Number of matches
- Filtering cutoff : 4
- Local residue angle deviation: 0.15 radians
- Short distance deviation: 0.3A
- Long distance deviation: 0.6A
By raising fuzziness you will likely retrieve more (many more) hits, but you'll also increase possible noise.
¶ Computational performance
On top of scientific accuracy, you typically want results as quickly as possible. So assessing how fast the search is, is also important. A full binding site search from 3decision will probably be a bit slower than a subpocket search (more heavy lifting on the database side). The more fuzzy you put your parameters, the more results you'll retrieve and have to postprocess. This will also impact performance. For typical full pocket search, 3decision's pocket comparison should take around a minute and for subpocket searches usually less.