¶ Introduction
Interaction search is a feature to retrieve structures containing a 3D query pattern of interactions. The matching is based on a selection of interactions in a query system. The user can refine interactants and interaction type, interaction length, and orientation tolerance. The returned hits are superimposed on the query and can be displayed in the 3D viewer.
¶ Cautions
This search handles a wide amount of 3D data, and a fuzzy search can last tens of minutes. A good practice is to start with a precise query as a first search, and then to make it progressively fuzzier if the number of hits is too limited.
¶ Interaction search feature access
The interaction search tool is enabled when selecting the Interaction Search option in the 3D viewer menu, when at least one interaction is selected.
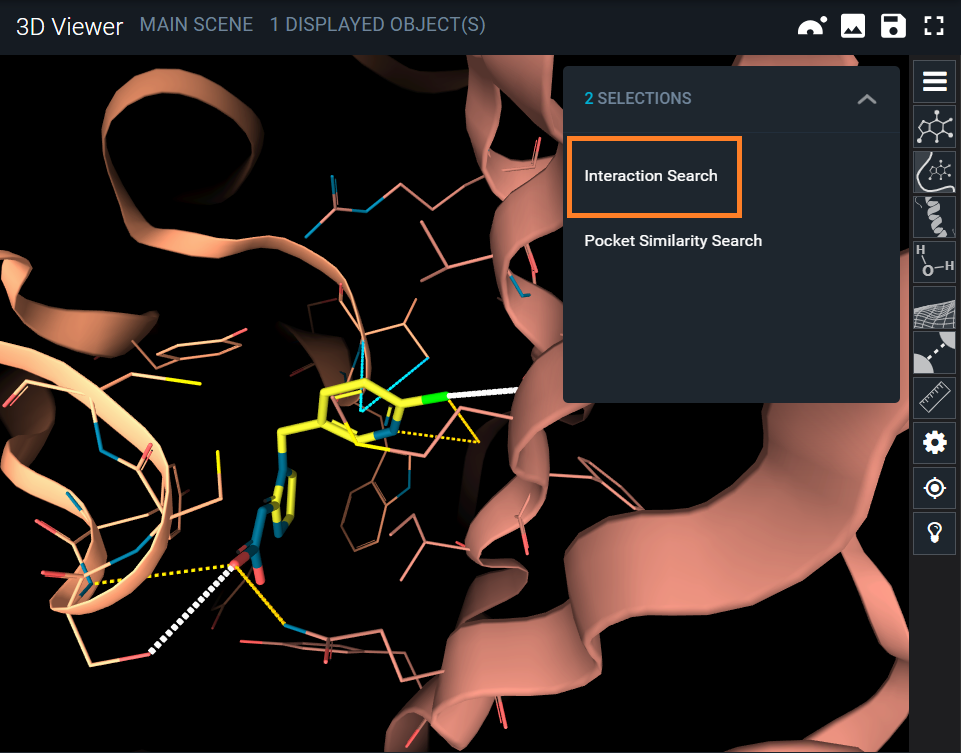
From the Interaction Search panel, you will be able to customize your query.
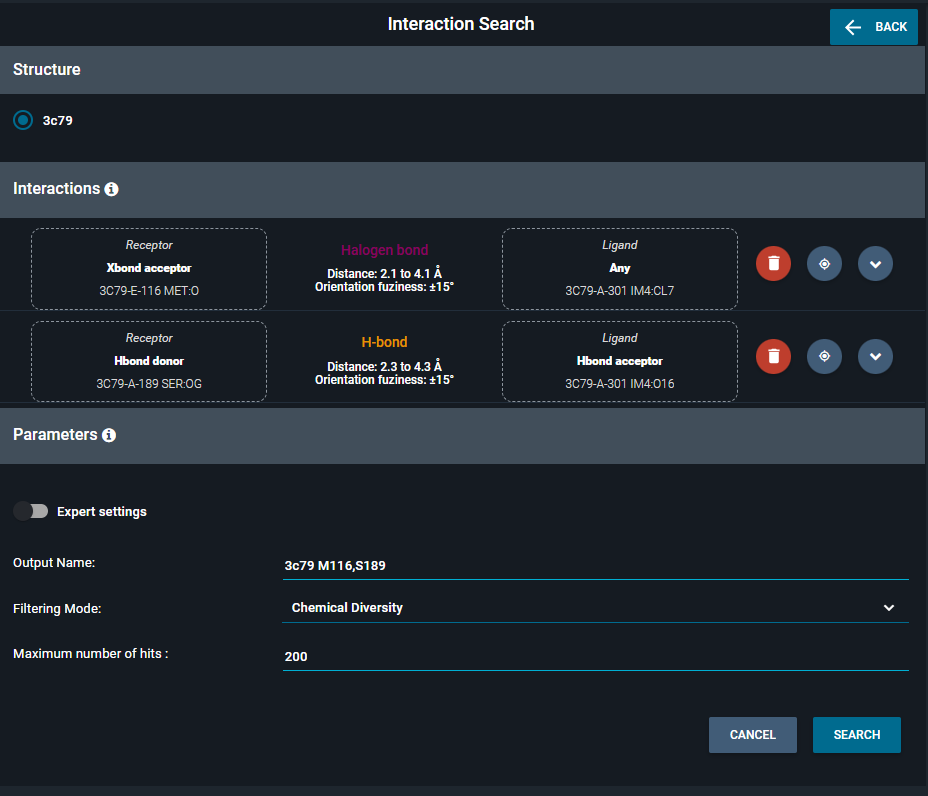
¶ Search parameters and algorithm
¶ Query
A query is a set of vectors, each one having a ligand atom type, a receptor atom type, and a vector type. Two additional parameters are provided for the query: the vector length range, and the orientation fuzziness. An example of an interaction query would be a H-bond edge between a ligand H-bond donor and a receptor H-bond acceptor, with a length between 2.1 and 4.1A and an orientation fuzziness of 15°. A search requires at least two vectors as a query.
¶ Hit
A hit structure will contain interactions matching all the query vectors. A partial match is not possible with the current algorithm. Hit structure must have matching interactions for each query vector. Each hit vector must match the query criteria:
-
ligand interactant type
-
receptor interactant type
-
interaction distance
Additionally, the 3D combinations of each pair of interactions are compared to the geometry of the corresponding query pair:
-
ligand interactants have a similar distance between them (default tolerance is 1Å)
-
receptor interactants have a similar distance between them (default tolerance is 1Å)
-
the angle between the 2 vectors is similar (tolerance is given by the highest 'orientation fuzziness' parameter out of the two vectors). Example: "2 H-bonds with an angle of 22°" between them can match with a query "2 H-bonds with an 8° angle and orientation fuzziness of 10° and 20°", as the angle difference of 14° is below the permitted 15°.
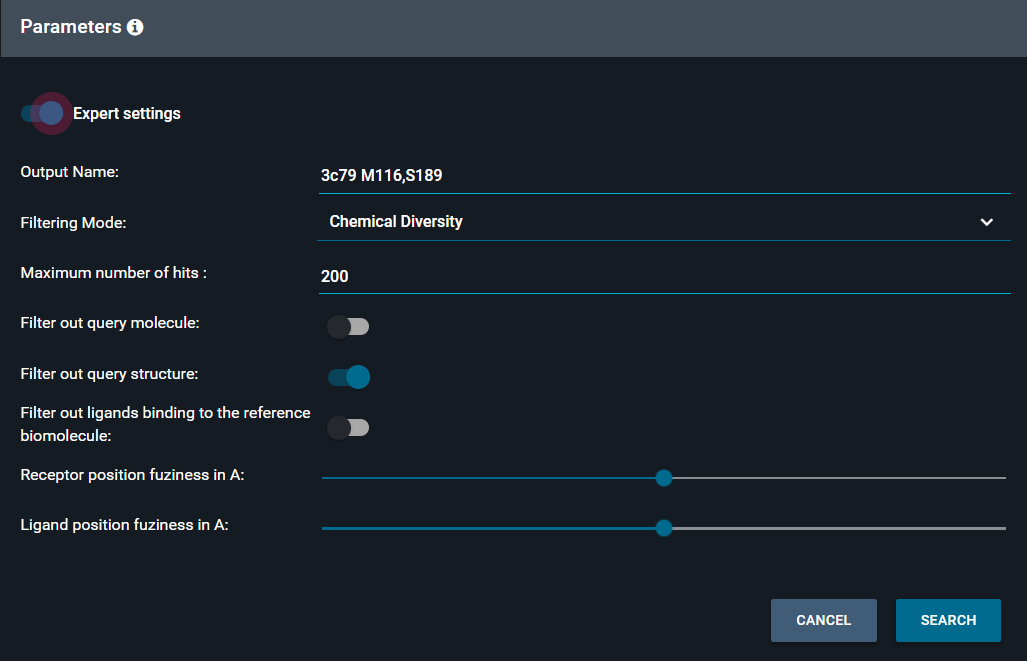
¶ Parameters
The selection of interactions in the 3D Viewer will prefill the interaction query with default parameters. The user can then refine the query.
For each interaction, the user can customize:
-
interaction type
-
interaction length
-
orientation fuzziness
-
interactant types
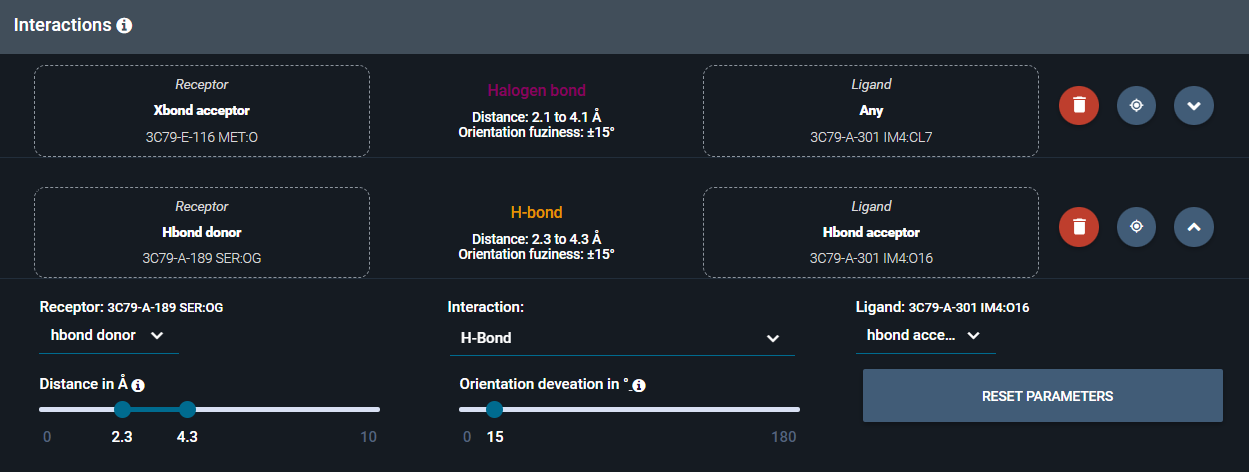
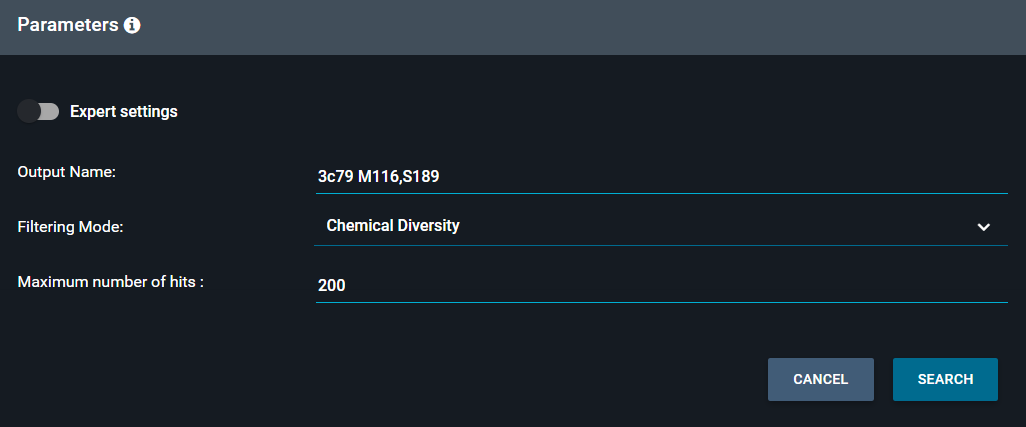
Additionally, some global parameters are available to filter the hits:
-
a limit can be applied to the number of hits (default is 200), to avoid excessive processing time for fuzzy queries
-
different filtering modes are available:
-
Chemical Diversity: one structure only is kept for each ligand
-
Biomol Diversity: one structure only is kept for each receptor biomolecule
-
Display All Hits
-
You can also customize the output name, to set the name of the collection of hits that will be displayed in the Workspace.
¶ Expert settings
To fine tune your search settings, you can toggle on the Expert settings and modify the fields:
-
Filter out query molecule: when toggled one, it removes the ligand that is in the reference structure you used as query
-
Filter out query structure: when toggled one, it removes the structure from the output collection (on by default)
-
Filter out ligands binding to the reference biomolecule: when toggled one, it removes structures containing the same biomolecule as the reference structure from the output collection
-
Receptor position fuzziness in Å (default is 1Å)
-
Ligand position fuzziness in Å (default is 1Å)
¶ Scoring
The scoring is based on a RMSD of interactants coordinates, after superimposition. Only ligand atoms and receptor atoms involved in matching interactions are used.
Formula used is Score = 1 - RMSD
The RMSD is transformed to have a score going with 1 as perfect match and lower values for less exact match. A value of 0 would mean that hit atoms have a RMSD of 1 compared to query, and negative values will indicates RMSD higher than 1A. We usually consider that negative values reports bad matches, but depending of the type of use case, a significant RMSD could be tolerated (like for scaffold hopping).
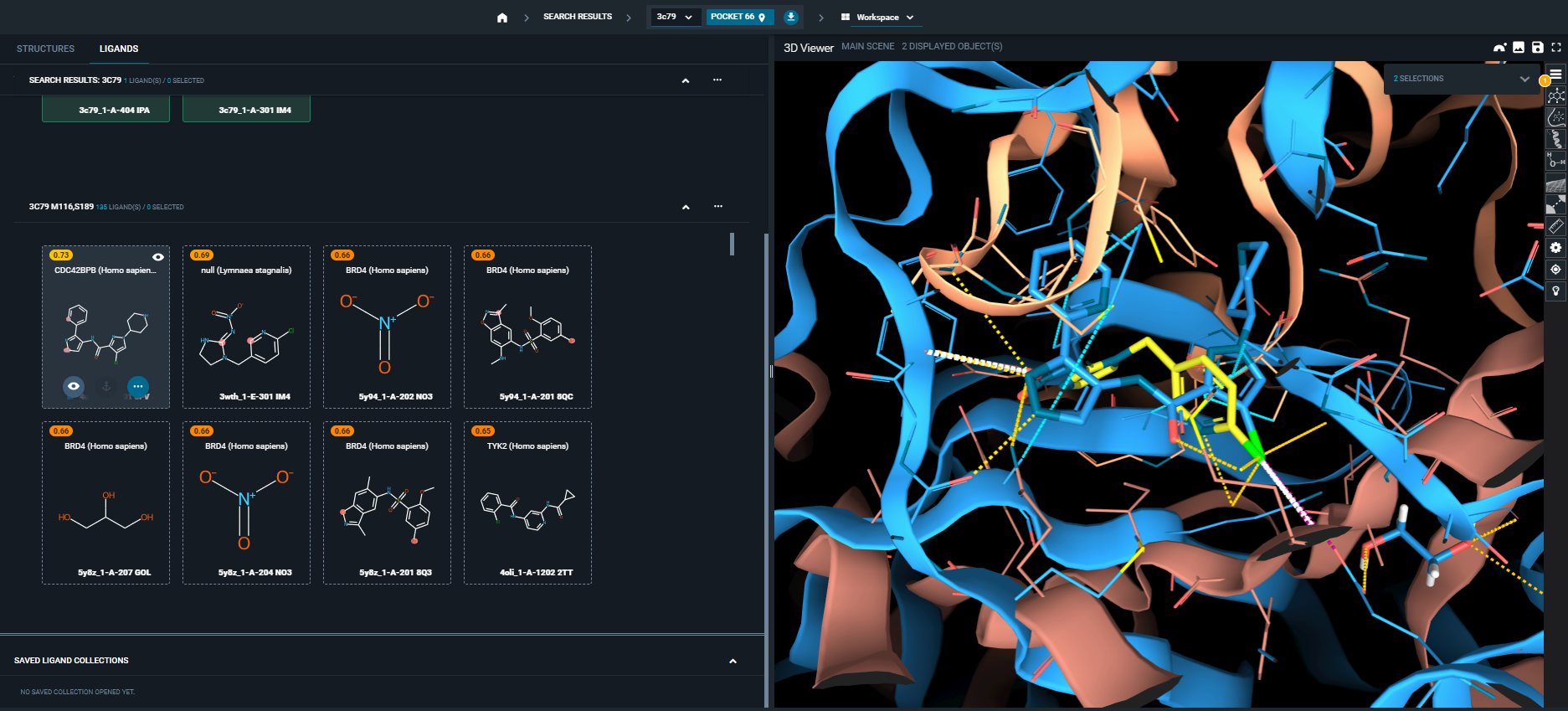
¶ Hits display
Hit ligands are displayed as ligand cards, sorted by score (a higher score means best hit). The hit ligands atoms that match the query are indicated in a red sphere. Moving the mouse over the card will display the eye icone, which allow the structure display. The hit structure is then displayed in 3D viewer, superimposed to the query using matching interactions only.
¶ Interactant types
Interactants are atoms (or atom groups) involved into a ligand-receptor interaction. In order to define contacts as interactions, interactants were categorized based on their connectivity, hybridization and partial charge. Each interactant can hold one or multiple types.
Interactant types used are :
- Anion
- Atom negatively charged.
- Apolar
- Atom with a neglectable partial charge.
- Aromatic
- Non-nitrogen atom that is part of an aromatic system.
- Aromatic N
- Nitrogen atom that is part of an aromatic system.
- Carbonyl carbon
- Carbon from a carbonyl function.
- Carbonyl oxygen
- Oxygen from a carbonyl function.
- Cation
- Atom positively charged.
- Function - amide
- Carbon or nitrogen atom from an amide function.
- H-bond acceptor
- Atom able to participate as hydrogen acceptor in an hydrogen bond.
- H-bond donor
- Atom able to participate as hydrogen donor in an hydrogen bond.
- Hydrophobic
- Hydrophobic atom.
- Metals
- Metalic atom.
- Neg ionisable
- Negatively ionisable atom.
- Polar
- Atom with a non-neglectable partial charge.
- Pos ionisable
- Positively ionisable atom.
- Uncharacterized
- Atom with uncharacterized interacting capabilities.
- Water
- Oxygen from water molecule.
- Weak H-bond acceptor
- Atom able to participate as hydrogen acceptor in a weak hydrogen bond.
- Weak H-bond donor
- Atom able to participate as hydrogen donor in a weak hydrogen bond.
- X-bond acceptor
- Atom able to participate as halogen acceptor in an halogen bond.
- X-bond donor
- Atom able to participate as halogen donor in an halogen bond.
¶ Interaction types
Presently, inter-ligand interactions or interactions between receptor residues are not taken into account. The interactions are detected between ligand and the receptor atoms (residues or water molecules). Then, a typing is performed based on interactant types and geometry of the contact. The results are either an interaction type or an interaction type group. They are stored in the database and returned to display interactions as dotted lines in 3Dviewer, or as part of specific features like interaction search.
Interaction types and interaction type groups used are:
¶ Apolar
- Hydrophobic
- Interaction between hydrophobic interactants
¶ Polar
- Amide interaction
- interaction involving a plan amide function
- Halogen Bond
- interaction between an halogen and an atom with lone pair
- H-Bond
- interaction between an interactant typed as hydrogen bond donor and another typed as hydrogen bond acceptor
- Polar interaction
- interaction between polar interactants, one with a positive partial charge and the other with a negative partial charge
- Weak polar
- Interaction between a weak polar interactant and an aromatic side
- Water Mediated Interaction
- Polar interaction between water and receptor for water molecules involved in at least another contact with a ligand
¶ Electrostatic
- Ionic
- Interaction between anion and cation interactants
- Potential Ionic
- Interaction between negatively ionisable and positively ionisable interactants
¶ Aromatic
Classic π–π stackings geometry are handeled:
-
Aromatic Sandwich : refers to a strictly parallel interaction between two aromatic rings. In this type of contact, the two aromatic rings are stacked on top of each other, with their planes perfectly aligned. This arrangement is sometimes referred to as a pi-stacking interaction.
-
Aromatic T-shape: this interaction occurs between the side of one aromatic ring and the center of another. In this configuration, the two aromatic rings are perpendicular to each other, forming a T-shape.
-
Parallel-displaced Aromatic: in this type of contact, the aromatic rings are shifted but remain parallel to each other. This interaction is characterized by a displacement of the rings along their common axis, resulting in a non-stacked, parallel configuration.
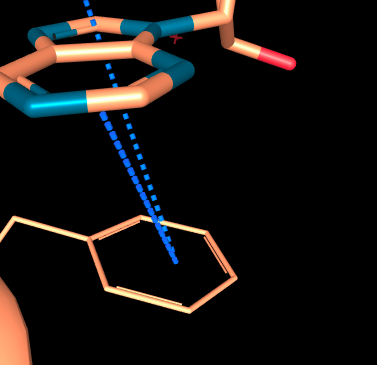
Additional interaction types were also added to manage further aromatic contacts in specific geometries, when angle are out of strict π stackings, parallel displaced or T-shape conformations:
- Aromatic Edge-to-face: this type of interaction occurs between the edge of one aromatic ring and the face of another. They are not perpendicular enough to be considered as a T-shaped configuration.
- Aromatic Face-to-face
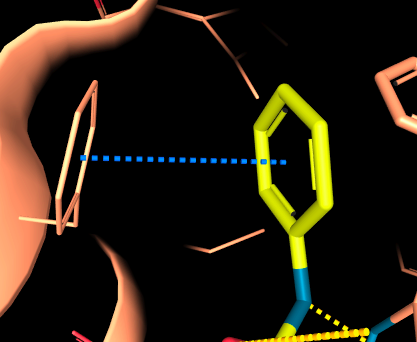
- Aromatic Side-by-side
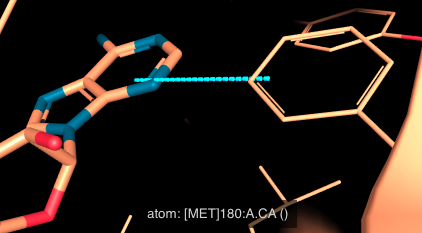
- Screw Aromatic: compared to a sandwich conformation, in a screw aromatic interaction, one ring is rotated around its axis, resulting in a screw-like arrangement.
The last type or aromatic contact imply an non armatic atom:
- Atom-Aromatic Face: in this interaction, a non-aromatic atom is in contact with the face of an aromatic ring.
¶ Suboptimal Contact
We used three categories of suboptimal contacts that can occur interactants. The first category involves contacts between polar and apolar interactants:
- Apolar contact with aromatic side
- Proximity of an apolar atom with an aromatic side, with a geomatry that would satisfy a polar-aromatic face contact.
- Hydrophobic Polar Contact
The second category of suboptimal contacts is characterized by non-complementary electrostatic charges. These interactions are referred to as ionic suboptimal contacts. They can be considered as potential ionic interactions, if one of the interactants is not charged for sure, ie if it can be in a charged state depending on the local environment or pH.
- Ionic Repulsion
- Unfavorable short range electrostatic interaction beetween charged atoms
- Potential Ionic Repulsion
- Potential unfavorable short range electrostatic interaction beetween a charged and a potentialy charged atom
The third category of suboptimal contacts involves low-distance contacts between atoms. This can result in a light clash, where the atoms are slightly clashing, and the distance between them is too short, leading to slightly unfavorable energies. In more severe cases, the atoms may be in critical clash, indicating a real clash between them. Additionally, if the distance between two atoms falls within a range that could allow for a covalent bond, the contact is registered as a potential covalent bond, although no chemical rules or filtering are applied, only distance criteria:
- Light Clash
- Atoms involed are slightly clashing : the distance between these atoms is too short and will result in slightly unfavorable energies
- Critical Clash
- involved atoms are to close, indicating a real clash between them
- Potential Covalent Bond
- when distance between 2 atoms in a range that could match with a covalent bond, the contact is registered as potential covalent bond. No chemical rules or filtering is applied here, only distance criteria.
¶ Uncharacterized
- Uncharacterized
- Interactions involving one or more uncharacterized interactant
- Uncharacterized Aromatic
- Interactions involving aromatic-aromatic interaction with undefined geometry
In Interaction search module, it is possible to search for any interaction type from a group. To do so, select the 'Any' interaction type associate to the group. For exemple, select the type 'Any Polar' will allow to map the interaction to any type from the group 'Polar' (i.e. Amide interaction, H-Bond, Halogen bond ...).