¶ Overview
The Highlight Mode is intended for comparing closely related structures where differences are subtle but biologically relevant. While changes in ligand conformation are usually easy to identify using standard visualization tools, small rearrangements in the protein—particularly in and around the binding site—are often difficult to detect visually.
Highlight Mode simplifies this analysis by allowing you to highlight, in a single click, all residues that differ from a reference structure. Differences can be based on sequence variations or on side‑chain movements exceeding a user‑defined RMSD threshold, making it immediately clear which regions have actually changed. Highlight Mode also ensures that incomplete residues, construct differences, and atom‑level discrepancies are explicitly captured during comparison.
Typical use cases include:
- Identifying ligand‑induced local rearrangements of residues in the binding site.
- Comparing different constructs of the same protein to reveal construct‑dependent variations.
- Comparing homologous proteins across species to reveal binding‑site differences.
- Identifying point mutations and amino‑acid substitutions that alter binding‑site geometry or physicochemical properties.
Contents
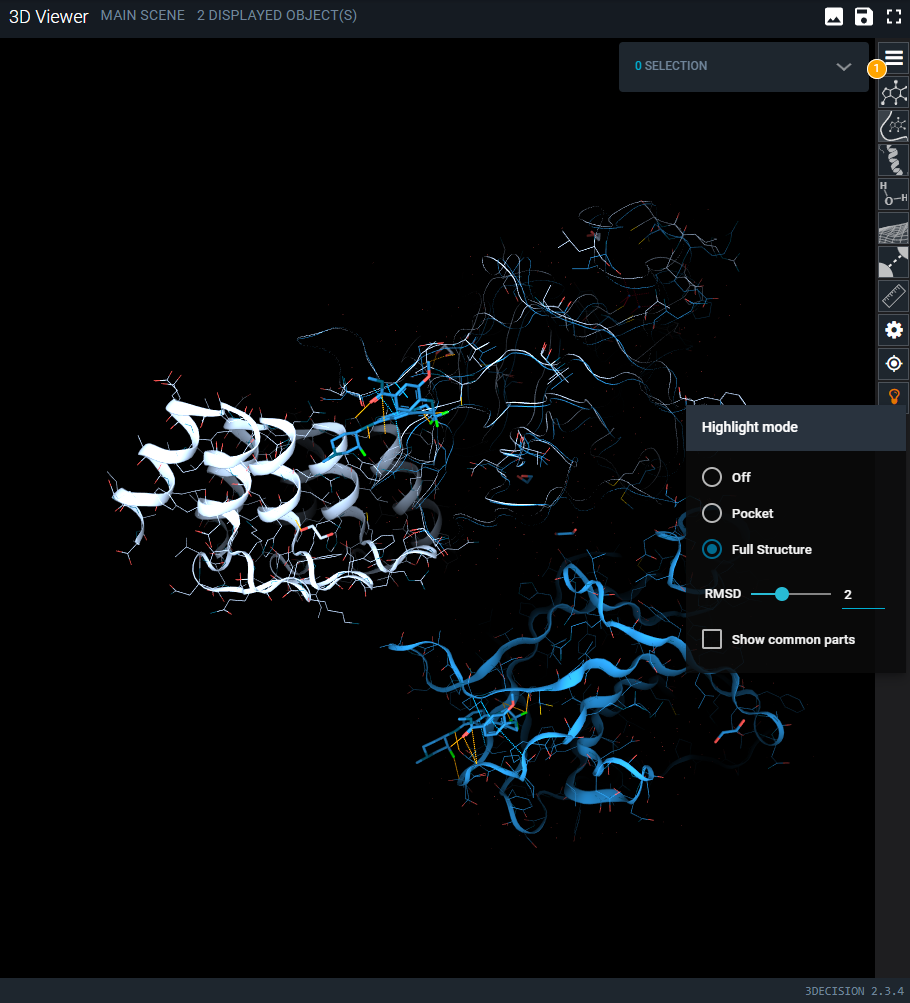
¶ Highlight Mode Menu
The bulb icon on the side toolbar opens the Highlight Mode menu. There are 2 different modes:
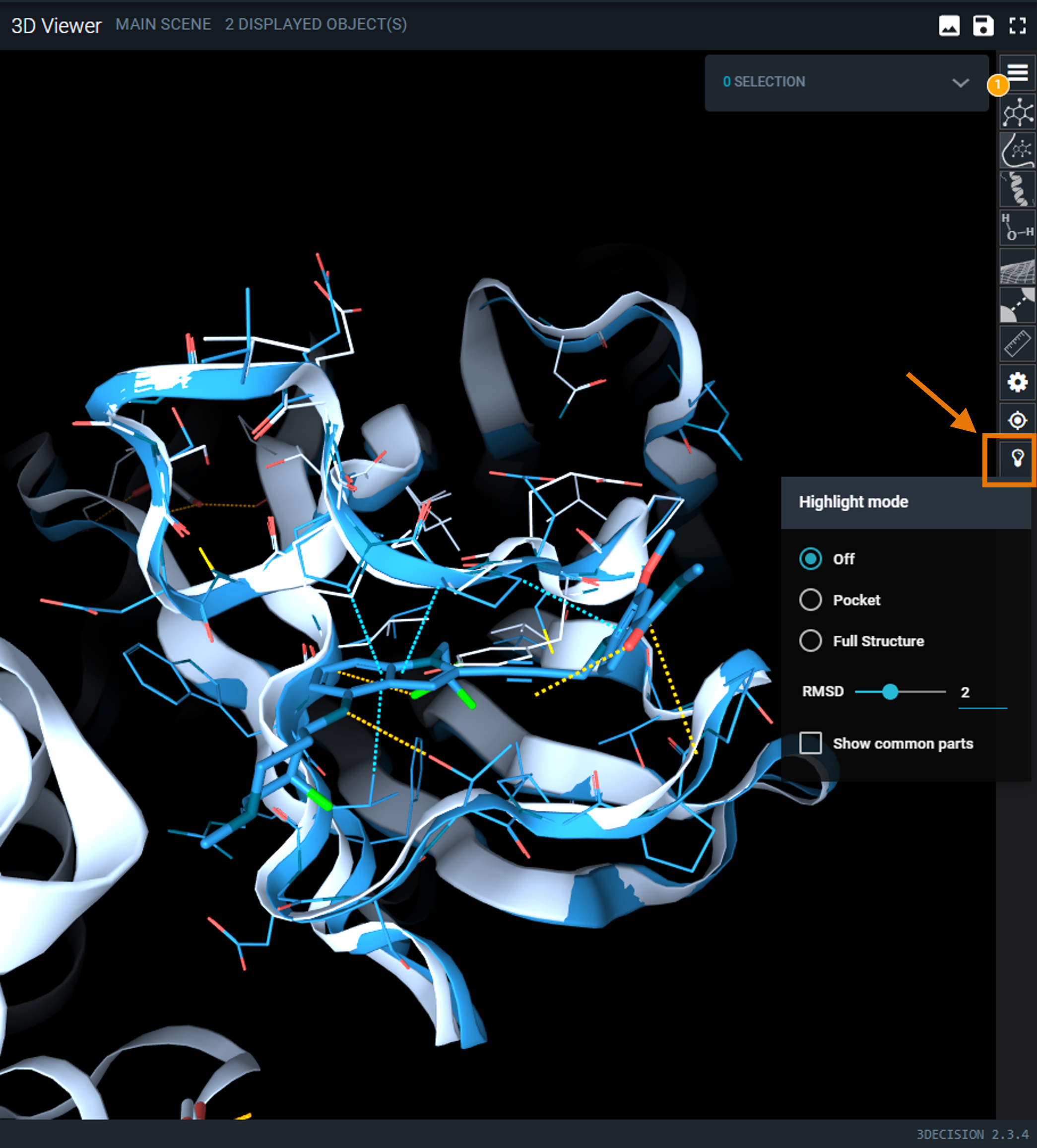 |
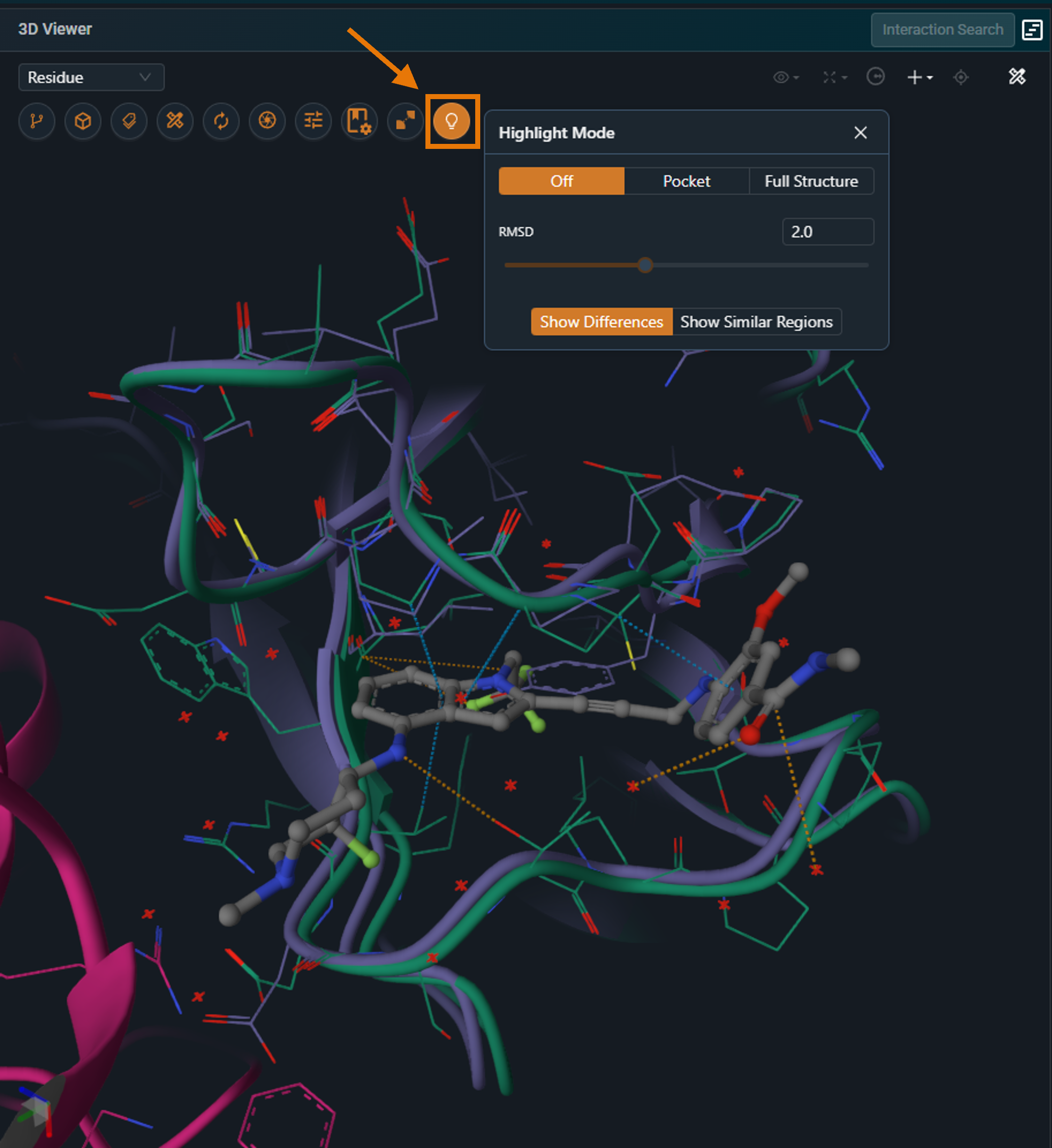 |
|---|
This mode focuses on the binding site and operates as described below only when the Workspace has a reference pocket. If no reference pocket is defined, the highlight behavior is identical to the Full Structure option.
The pocket mode is useful when studying binding site differences across targets and off-targets or comparing different conformations of the same target. It will display differences within the superposed pockets. Residues that differ between the structures are displayed using line and cartoon representations, while residues that are identical are hidden. The full protein chains remain visible as a thin putty representation to provide structural context. In addition, a surface for the reference binding site.
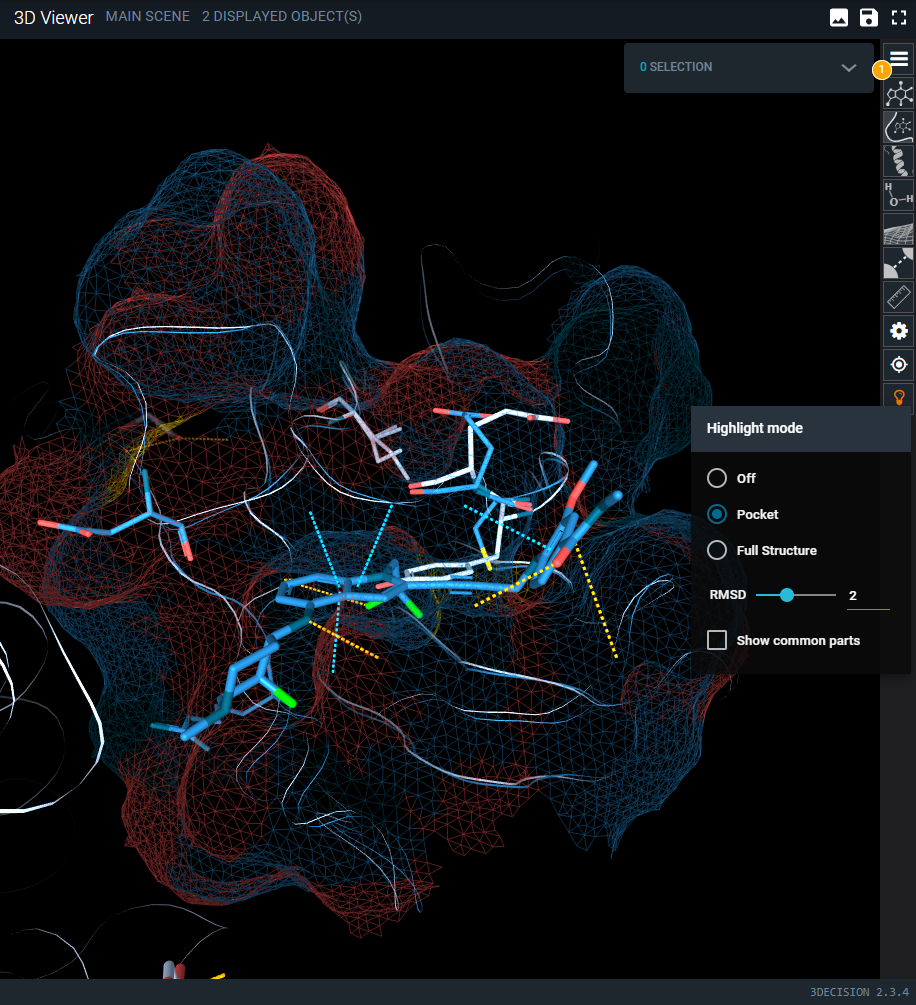 |
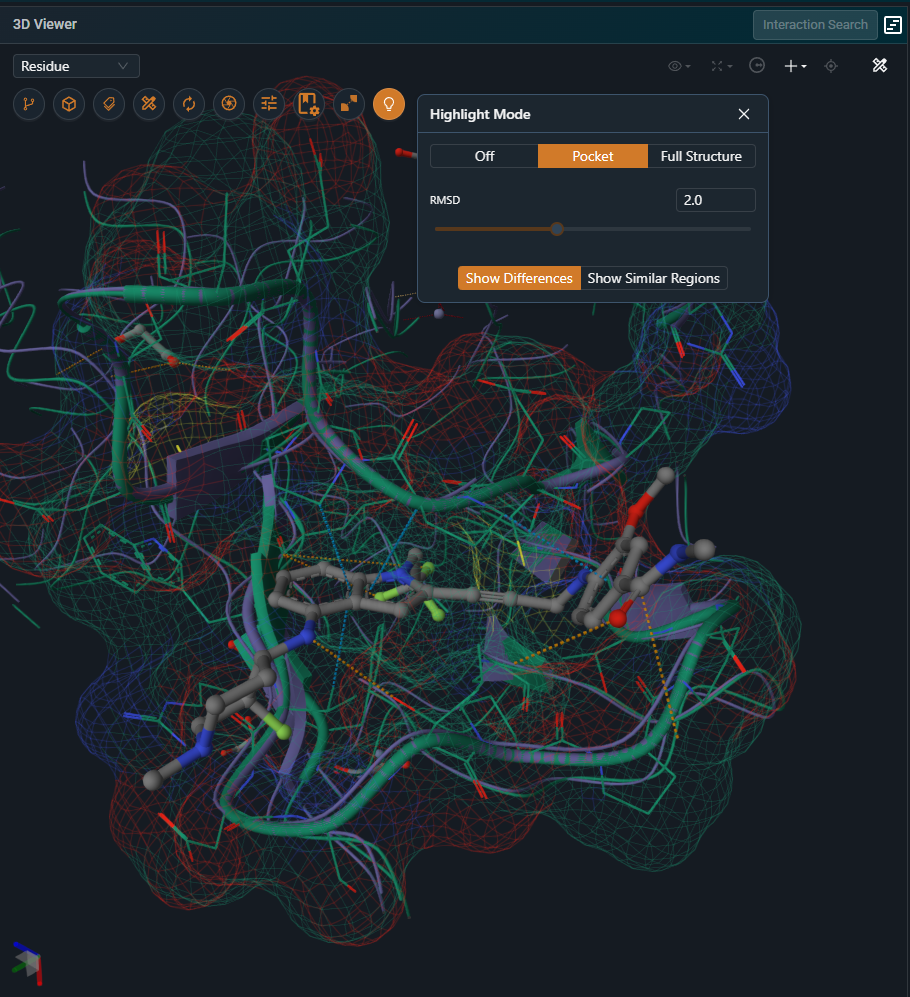 |
|---|
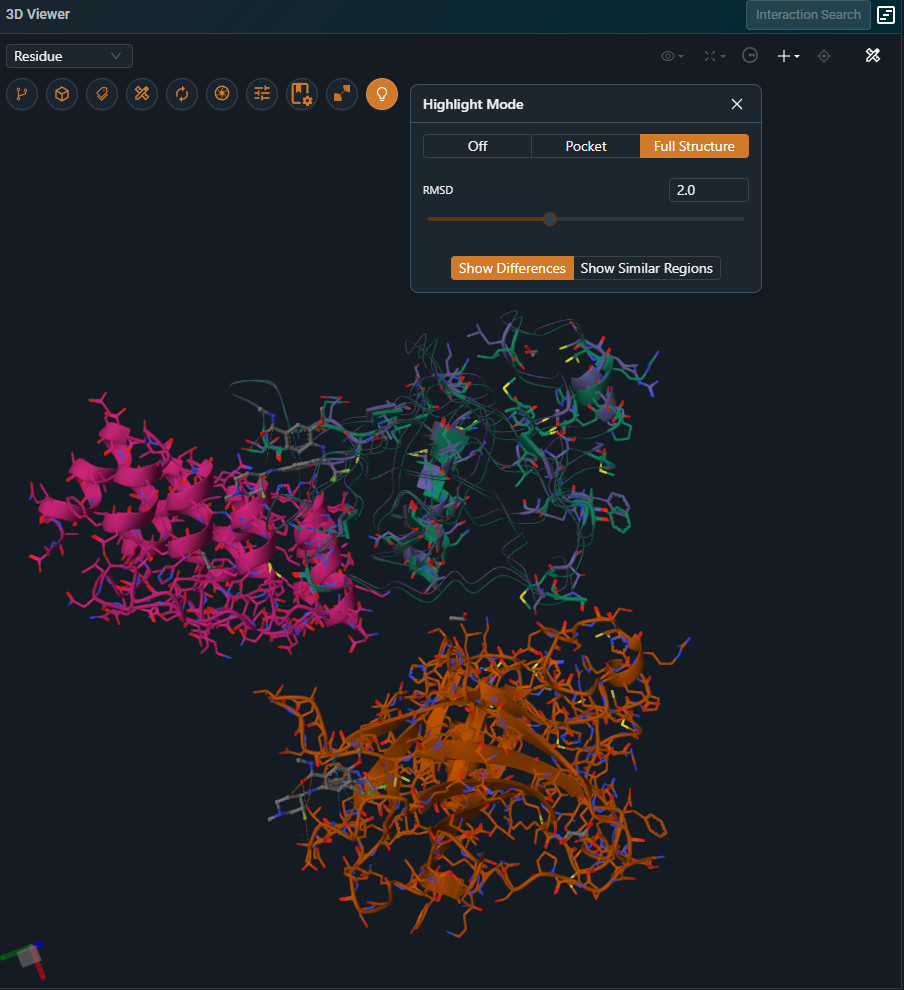
¶ Full Structure
This mode highlights residue‑level differences across the entire structure. Residues that differ between the two structures are displayed using line and cartoon representations, while residues that are identical are hidden. The full protein chains remain visible as a thin putty representation to provide structural context.
Keep in mind that the structural overlay is optimized on the region used for the alignment, which can be either the pocket or a selected chain. As a result, Highlight Mode is most reliable for interpreting subtle differences within that aligned region. Outside of it, residues may appear as shifted simply because they are not part of the superposed area and therefore fall outside the scope of the comparison.
 |
 |
|---|
¶ Other Options
The RMSD slider allows you to control the RMSD threshold used to compare matching residues. If the side-chain RMSD between two matching residues is higher than the selected threshold, that residue is treated as a difference.
The highlight mode can also be inverted to display only residues that are similar between the compared structures. In the current user interface, this option is available as a checkbox labeled “Show common part”. In the new user interface, it is accessed via the “Show Similar Regions” button.
¶ How Highlight Mode Compares Residues
¶ How Residues Are Matched Across Structures
To determine which pairs of residues should be compared, Highlight Mode compares the positions of the Cα atoms of each structure against those of the Workspace reference structure. Residues whose Cα atoms fall within a 2 Å spatial distance are considered equivalent and are compared to identify similarities or differences. These are called matching residues.
¶ What Counts as a Difference
Once two residues are matched, they are compared in more detail. Residues will be highlighted as different in these scenarios
- Conformational change: the residue conformation differs beyond the defined RMSD threshold. Note: the representation highlights which part of the residue has moved—backbone differences are shown in cartoon, while side-chain differences are shown in licorice.
- Sequence variation: the residue codes are different.
- Missing atoms: the number of resolved atoms is different
- Missing resiudes: no matching residue is found — for example due to missing residues, missing loops, or large conformational differences.
Complete 'flips' of symmetrical atom groups, such as the aromatic ring in Phe and Tyr residues, or the carboxyl group in Glu and Asp, are not highlighted as differences.
¶ Special Cases and Exceptions
¶ Pocket Mode Limitation in the Legacy UI
In the legacy UI, Pocket Mode has a limitation: only residues present in both pockets are considered during residue comparison. Residues that are present in only one of the two pockets are automatically flagged as different, even though this may not reflect a true difference. This limitation has been addressed and corrected in the new UI.
¶ Reference Structure Definition
Highlight Mode always uses the workspace reference structure as the reference. The reference structure must be loaded in the 3D viewer for Highlight Mode to work.
To change the reference used by Highlight Mode, update the workspace reference and ensure that this structure is loaded and visible in the 3D viewer.